Det
21. Landsmøte i kjemi
Foredrag
- Abstracts
Fellesarrangemnetet står først,
ellers er foredragene nummerert etter faggruppe:
FE
- Fellesarrangement
AN
- Analytisk kjemi
KA
- Katalyse
HI -
Kjemiens historie
KI - Kjemometri
UN
- Kjemiundervisning
KM
- Kvantekjemi og
modellering
MK
- Makromolekyl- og
kolloidkjemi
MA
- Matkjemi
OR
- Organisk kjemi
UM
- Uorganisk kjemi og materialkjemi
Postere
Dette dokumentet oppdateres etterhvert som abstractene kommer inn.
FE -
Fellesarrangement
GW
Guldberg-Waage-foredrag
Hydrogen
Truls Norby
Kjemisk Institutt, Universitetet i Oslo
Starting
my first
research in inorganic chemistry, I stumbled across hydrogen where and
when I
and everyone else did not expect to find it – it is so common
and yet
so
un-common. It makes bonds more different than any other element
– polar
and purely
covalent as protons and atoms, metallic when it pleases, and ionic as
hydride
ions. Consequently, it shows up in very different forms and locations
and has taken
me for a journey to my roots in physical chemistry and to
electrochemistry –
some things discovered and some mysteries remaining. Hydrogen holds a
historical role in the establishment of major Norwegian industry, and
we today
host world-leading production of electrolyzers for the emerging markets
of
hydrogen for storage and as carrier of renewable electrical energy.
Many
different types
of solids contain or may take up water and become solid-state proton
conductors
by various mechanisms of transport. They may be used as electrolytes in
novel
types of fuel cells and electrolyzers for hydrogen and renewable
energy, as
well as in electrocatalytic reactors for upgrading natural gas to
liquids or
hydrogen with minimal carbon emissions or with carbon capture and
storage. The
research involves experimental and ab initio computational methods to
understand
hydrogen in its various forms from gas phase, via surfaces and charge
transfer
at electrode interfaces, into mobile ions in crystalline or liquid-like
condensed phases.
FE1
Bottom-up Assembly of Active, Autonomous and Complex
Bioinspired Systems with Adaptive Behaviour
Daniela Wilson
Systems Chemistry, Radboud University Nijmegen, Institute for
Molecules and Materials Nijmegen, The Netherlands
d.wilson@science.ru.nl
Self-powered artificial motile systems are currently attracting
increased interest as mimics of biological motors but also as potential
components of nanomachinery, robotics, and sensing devices [1]. We have
recently demonstrated a supramolecular approach to design synthetic
nanomotors using self-assembly of amphiphilic block copolymers into
polymersomes and the controlled folding of the vesicles under osmotic
stress into a bowl shape morphology [2]. The folding process can be
precisely controlled to generate different complex architectures [3]
with adjustable openings and selective entrapment of inorganic
catalysts [4,5] enzymes or multiple enzymes working together in a
metabolic pathway [6,7]. Control of the speed and behaviour of the
nanomotors is possible due to integration of regulatory feedback and
feedforward loops in the enzyme networks designed to preserve energy
and run the motors at even lower concentrations of fuel eg. 0.05 mM
Glucose. Movement in both blood serum and plasma at physiological
concentrations of substrates is consequently demonstrated. The
nanomotor is now not only running at low concentrations of fuel but
also able to regulate it's fuel consumption to achieve the same output
speed showing adaptive behaviour. Recent developments on greater
control over the movement of the nanomotors under chemical gradients or
temperature will be presented [4,7]. Additional manipulation of the
nanomotors under external stimuli and their biomedical applications
will be discussed [6,7].
Acknowledgement
This work was supported in part by the European Research Council under
the European Union's Seventh Framework Programme
(FP7/2007-20012)/ERC-StG 307679 (StomaMotors).
References
- a) Abdelmohsen, L. K. E. A., Peng, F., Tu, Y. Wilson, D.
A., J.
Mater. Chem. B., 2014,
2, 2395-2408. (b) Tu, Y. Peng, F. Adawy, A. Men,
Y.; Abdelmohsen, L.K.E.A.; Wilson, D. A. Chem. Rev. 2016, doi:
10.1021/acs.chemrev.5b00344 c) Fei Peng, Yingfeng Tu, Daniela A. Wilson
Chem. Soc. Rev. 2017,
DOI: 10.1039/C6CS00885B
- (a) Wilson, D.A., Nolte, R, J. M., van Hest, J.C.M. Nature
Chem. 2012,
4,
268-274. b) Wilson, D.A., Nolte, R, J. M., van Hest, J.C.M. J.
Am. Chem. Soc., 134,
9894, (2012). (b) Wilson, D.A., de Nijs, B., van
Blaaderen, A., Nolte, R, J. M., van Hest, J.C.M., Nanoscale, 2013, 5,
1315.
- (a) R.S.M. Rikken, H. Engelkamp, R.J.M. Nolte, J.C. Maan,
J.C.M.
van Hest, D.A. Wilson& P.C.M. Christianen "Shaping
polymersomes into predictable morphologies via out-of-equilibrium
self-assembly", Nat. Commun 2016,
doi:10.1038/NCOMMS12606 (b)
Fei Peng, Nannan Deng, Yingfeng Tu, Jan C.M. van Hest, Daniela A.
Wilson, Nanoscale 2017
DOI: 10.1039/C7NR00142H (b)
- a) Abdelmohsen, L. K. E. A., Nijemeisland, M, Pawar, G. M.
Janssen, G.-J. A. Nolte, R. J. M., van Hest, J. C. M.
&
Wilson, D.A. *, ACS Nano, 2016,
10 (2), pp 2652-2660. b) Peng, F.
Tu, Y. Pierson, L., van Hest, J. C. M., Wilson, D. A.*, Angew. Chem.
Int. Ed. 2015,
54 (40) 11662-11665
- a) R. Rikken, R.J.M. Nolte, J.C. Maan, J.C M van Hest, D.
A.
Wilson P.C.M. Christianen, Soft Matter, 2013, DOI:
10.1039/C3SM52294F
R. Rikken, R.J.M. Nolte, J.C. Maan, J.C M van Hest, P.C.M. Christianen,
D. A. Wilson Chem Commun, 2013,
DOI:10.1039/C3CC47483F
- a) Rhee, P. G.; Rikken, R. S.; Nolte, R. J. M., Maan, J.
C., van
Hest, J. C. M., Christianen, P. C. M. and Wilson, D. A.* Nature Commun.
5, 2014,
doi: 10.1038/ncomms6010. b) Fei Peng, Yingfeng Tu, Jan C.M.
van Hest, Wilson, D. A.*, Adv. Mater., 2016, DOI:
10.1002/adma.201604996.
- (a) Yingfeng Tu, Fei Peng, Xiaofeng Sui, Paul White, Jan
C.M. van
Hest, Wilson, D. A. Nature. Chem. 2017
DOI: 10.1038/nchem.2674. (b)
Yingfeng Tu, Fei Peng, Alain Andre, Yongjun Men, Daniela A. Wilson*,
ACS Nano 2017, DOI:10.1021/acsnano.6b08079 (c) Fei Peng, Yingfeng Tu,
Ashish Adhikari, Jordi J.C.J Hintzen, Dennis Lowik, Daniela A. Wilson*
Chem Commun 2017, 53, 1088-1091. (d) Fei Peng, Yongjun Men, Yingfeng
Tu, Daniela A. Wilson, Adv. Funct. Mater. 2018, 10.1002/adfm.201706117
(e) Yingfeng Tu, Fei Peng, Paul B. White, Daniela A. Wilson, Angew.
Chem. Int. Ed. 2017, doi: 10.1002/anie.201703276, 56 (26), 7620-7624
FE2
Molecular Assemblers, Molecular Machines Performing Synthesis
R. Herges
Otto-Diels Institute for Organic Chemistry, University of
Kiel, Germany
In chemical synthesis usually the reactants are dissolved in an organic
solvent, the reactive molecules undergo stochastic collisions and form
a bond if kinetic energy and relative orientation are favourable.
However, the majority of biologically active molecules in nature are
synthesized in ATP driven, molecular machine-type enzyme complexes such
as non-ribosomal peptide synthetases (NRPS) or polyketide synthases
(PKS). They operate like an assembly line by guiding reactions under
positioning control driven by ATP. Notwithstanding the fact that there
are a number of advantages to this assembler-like synthesis (less side
reactions, easy stereo control, no protecting groups, preselection of
reactants, driving unfavourable reactions…), there is no
artificial system published so far.
We are aiming at the design, synthesis and investigation of the first
model system of a molecular assembler. In our preliminary work we
designed and synthesized a light-switchable ditopic receptor which is
able to drive the condensation of 4 molecules of vanadate to a cyclic
tetravanadate. The reaction which is endergonic and therefore not
spontaneous in the absence of the ligand is driven by the large and
selective binding energy of the product tetravanadate inside the
receptor. Photochemical isomerization (365 nm) of the ligand releases
the product. Upon irradiation with 430 nm the original,
“empty” state is restored and the cycle starts again.
FE3
Industry lecture
Radionuclides
and cancer treatment: How to succeed
Roy Larsen
Oncoinvent AS
Radionuclides have been used for cancer
treatment for almost a century.
Initially gamma and beta emitters were used but later alpha emitters
attracted
a substantial attention. Criterion for successful product development
should be
determined before initiation of the clinical phase of product
development. The
product candidate’s chemical and physical properties must be
carefully
considered, and synthesis route should be adaptable to industrial
scale. The
product candidate must show consistent antitumor activity and
acceptable safety
profile in the preclinical tests and dosimetry estimates for human use
should
indicate appropriate benefit to risk ratio. Sufficient patent
protection is
needed to attract investors.
Radionuclides and properties are addressed, and
examples of clinical
products are presented.
Norwegian inventions in the field are presented
and the international
trends in the field are discussed.
FE4
Integrating cryogenic ion chemistry and spectroscopy: Capture
and
characterization of reaction intermediates in homogeneous catalysis
Mark Johnson
Yale Univerisity
The coupling between ambient ionization sources, developed for mass
spectrometric analysis of biomolecules, and cryogenic ion processing,
originally designed to study astrochemistry, creates a new and general
way to capture transient chemical species and elucidate their
structures with optical spectroscopies. Advances in non-linear
optics over the past decade allow single-investigator, table top lasers
to access radiation from 550 cm-1 in the
infrared to the vacuum
ultraviolet. When spectra are acquired using predissociation of weakly
bound rare gas "tags", the resulting patterns are
equivalent to absorption spectra and correspond to target ions at
temperatures below 10K. Taken together, what emerges is a new and
powerful structural component to traditional mass spectrometric
analysis. Recent applications ranging from the mechanisms of
small molecule activation by homogeneous catalysts to the microscopic
mechanics underlying the Grotthuss proton relay mechanism in water
emphasize the generality and utility of the methods in contemporary
chemistry.
FE5
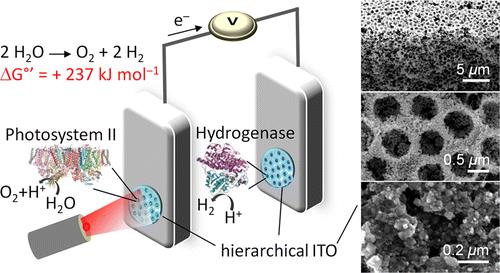
Semi-artificial Photosynthesis
Erwin Reisner
In photosynthesis, light is used for the production of chemical energy
carriers to fuel biological activity and the water oxidation enzyme
Photosystem II is the first protein complex in the light-dependent
reactions of oxygenic photosynthesis. This presentation will summarise
our progress in the development of protein film photoelectrochemistry
as a technique for the activity of this enzyme adsorbed onto an
electrode surface to be studied.[1] Materials design enabled us to
develop 'tailor-made' 3D electrode scaffolds for optimised
integration of the 'wired' enzyme and these investigations
yielded valuable insights into Photosystem II function. Examples are
the identification of unnatural charge transfer pathways to the
electrode and the elucidation of O2 reduction
pathway that
short-circuit the known water-oxidation process.[2]
The integration of Photosystem II in a photoelectrochemical circuit has
enabled the in vitro re-engineering of natural photosynthetic pathways.
We assembled an efficient semi-artificial water splitting cell driven
by light through the rational wiring of Photosystem II to a H2
producing enzyme known as hydrogenase (Figure 1).[3] This hydrogenase
displays unique properties for water splitting applications as it
displays good H2 evolution activity, little
product (H2) inhibition and
some tolerance towards O2.[4] The bio-hybrid
water splitting cell shows
how we can harvest and utilise electrons generated during water
oxidation at Photosystem II electrodes for the generation of renewable
H2 with a wired hydrogenase through a direct
pathway unavailable to
biology. Progress in the assembly of bias-free tandem water splitting
cells with wired enzymes and the integration of robust live
cyanobacteria in 3D structured electrodes will also be discussed.[5]
Figure 1. Schematic
representation of a semi-artificial water splitting system. Water is
photo-oxidized and O2 generated at a Photosystem II-containing
photoanode and aqueous protons are reduced at a hydrogenase-based
cathode. Enzyme-integration was optimised by using a hierarchical ITO
architecture.
References
- Kato, Zhang, Paul & Reisner, Chem. Soc. Rev., 2014,
43, 6485-6497.
- Zhang, Sokol, Paul, Romero, van Grondelle, &
Reisner, Nature Chem. Biol., 2016,
12, 1046-1052.
- Mersch, Lee, Zhang, Brinkert, Fontecilla-Camps, Rutherford
& Reisner J. Am. Chem. Soc., 2015, 137, 8541-8549.
- Wombwell, Caputo & Reisner, Acc. Chem. Res., 2015,
48, 2858-2865.
- Zhang, Bombelli, Sokol, Fantuzzi, Rutherford, Howe
& Reisner, J. Am. Chem. Soc., 2018, 140, 6-9.
AN -
Analytisk kjemi
AN1
ISO/IEC 17025:2017 - en oppdatert versjon av verdens mest
brukte
akkrediteringsstandard som omfatter generelle krav til
prøvings-
og kalibreringslaboratoriers kompetanse
Oversikt over endringer og likheter sammenlignet med ISO/IEC
17025:2005.
Maarten Aerts
Norsk
Akkreditering
Foredraget
er rettet mot laboratorie-ansatte som ønsker å
lære
mer om ISO/IEC 17025:2017 som kompetansestandard.
Akkrediteringsansvarlig i Norsk akkreditering, Maarten Aerts, vil
gjennomgå den nye oppbyggingen og strukturen av ISO/IEC
17025:2017, forklare hensikten og filosofien bak den nye versjonen,
samt gjennomgå noen sentrale kravelementer som er oppdatert
siden
2005-versjonen av standarden.
AN2
Validering av metode - Hvorfor, hvordan og
når er det nødvendig?
Elin
Lovise Folven Gjengedal
Norges miljø- og biovitenskapelige universitet,
Fakultet for miljøvitenskap og naturforvaltning, Ås
Hva er bakgrunnen og begrunnelsen for metodevalidering? Eurachem Guiden
“The Fitness for Purpose of Analytical Methods – A
Laboratory Guide to Method Validation and Related Topics”
forklarer hvorfor, hvordan og når validering av en
analysemetode
er nødvendig [1]. Foredraget vil handle om arbeidet med
guiden,
hvordan et valideringsstudium bør utføres og hvor
mye som
skal inngå i arbeidet (validering/verifisering), forklaring
på de ulike valideringsparameterne, oppfølging av
valideringsstudien og dokumentasjon på analysemetoden.
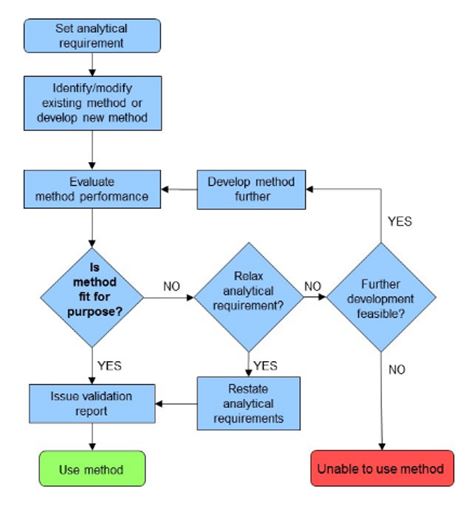
Figur 1.
Metodevalideringsprosessen [1]. Metodevalidering består av et
studium hvor ulike valideringsparametere blir vurdert og deretter
sammenlignet med analytiske krav. Metodens egnethet bestemmes av
hvordan metoden utfører når den utpekte
analytikeren
bruker det tilgjengelige utstyret/fasilitetene.
Referanser
- B. Magnusson and U. Örnemark (eds.) Eurachem
Guide: The
Fitness for Purpose of Analytical Methods – A Laboratory
Guide to
Method Validation and Related Topics, (2nd ed. 2014). ISBN
978-91-87461-59-0. Available from http://www.eurachem.org.
AN3
Utfordringer ved bestemmelse av deteksjonsgrenser
Grethe Wibetoe
Kjemisk institutt, Universitetet i Oslo
Ved bestemmelse av analytter i sporkonsentrasjoner er det
nødvendig å etablere deteksjonsgrenser (LOD) for
analysemetodene. Metodens LOD er kanskje den valideringsparameteren som
har vært gjenstand for mest diskusjon gjennom tidene og er
vanskeligst å etablere – spesielt for komplekse
analysemetoder.
Det er flere tilnærminger til bestemmelse av LOD, men metoden
basert på å multiplisere standardavviket til
blankprøve med en konstant (LOD = k·Sblank)
er
etter hvert blitt den mest vanlige og anerkjente metoden - der det er
praktisk å anvende den. Metoden er for eksempel beskrevet i
Eurachems valideringsguide for analysemetoder fra 2014 [1].
Selv om utrykket for bestemmelse av LOD er enkelt, er det mange
spørsmål knyttet til den praktiske
gjennomføringen
– spesielt for å få en mest mulig
realistisk LOD.
Rapportering av resultater nær og under LOD er også
et tema
som trengs å diskuteres.
Presentasjonen vil gi en kort teoretisk bakgrunn for metoden for
bestemmelse av LOD, og forskjellige utfordringer for å kunne
bestemme en realistisk deteksjonsgrense for analysemetoden vil bli
diskutert.
Referanser
- B. Magnusson and U. Örnemark (eds.) Eurachem
Guide: The
Fitness for Purpose of Analytical Methods – A Laboratory
Guide to
Method Validation and Related Topics, (2nd ed. 2014). ISBN
978-91-87461-59-0. Available from http://www.eurachem.org
AN4
Analytical challenges in Forensic Toxicology
Veronica Horpestad
Liane
Division of Laboratory Medicine, Department of Forensic
Sciences, Oslo University Hospital
The Department of Forensic Sciences provides scientific based knowledge
at a high international level for use in criminal and civil law. At the
section for forensic toxicological analytics, biological samples mainly
received from the police are analyzed. High quality analytical methods
are required for the analysis of pharmaceuticals and drugs of abuse
within this discipline as the results may cause legal sanctions.
In this presentation method validation, measurement of uncertainty and
safety margins will be focused. Analytical challenges due to
development in the pharmaceutical and illicit drug market will also be
mentioned.
AN5
Kjemiske våpen – jakten på
bevis.
Bent-Tore
Røen
Forsvarets Forskningsinstitutt
Kjemiske våpen er innretninger som inneholder giftige
kjemikalier, med en mekanisme for å spre kjemikaliene i
lufta,
for eksempel i form av en bombe. På tross av stor
internasjonal
oppslutning om forbud mot bruk av kjemiske våpen har giftige
kjemikalier den senere tiden blitt brukt i stor skala i Syria og Irak,
samt i målrettede attentat mot enkeltpersoner i Malaysia og
Storbritannia. Slike hendelser anses som grove brudd på
folkeretten da de påfører store lidelser for dem
som blir
eksponert, ofte med dødelig utfall.
For å overvåke at den siste internasjonale avtalen
om
forbud mot kjemiske våpen blir respektert
(Kjemivåpenkonvensjonen av 1997), ble Organisasjonen for
forbud
mot kjemiske våpen (OPCW) opprettet. En av oppgavene til OPCW
er
å føre bevis i tilfeller der kjemiske
våpen har
blitt brukt, for å kunne stille aktørene til
ansvar for
sine gjerninger. OPCW gjennomfører inspeksjoner der blant
annet
bevismateriale blir samlet inn i form av jord, bygningsmaterialer,
klær m.m., eller biologisk materiale fra antatt forgiftede
personer.
For å kunne etterprøve
kjemivåpenkonvensjonen er
OPCW avhengig av laboratorier som kan analysere prøvene, og
som
tilfredsstiller deres krav til kvalitet og kompetanse. I tillegg
må laboratoriene kunne håndtere svært
giftige
kjemikalier på en sikker måte, og ha tilgang til
eller
være i stand til å syntetisere relevante
referansematerialer. Laboratoriene må hvert år
også
delta i kvalitetstester ved at de mottar prøver med ukjent
innhold, der alle relevante kjemikalier må finnes og
rapporteres
i henhold til gitte kvalitetskrav. OPCW har i dag et tjuetalls
designerte laboratorier, og Forsvarets forskningsinstitutts (FFIs)
laboratorium på Kjeller er i ferd med å
oppnå en slik
OPCW-designasjon.
AN6
Pollutants in the Arctic.
Roland
Kallenborn1, Simon Wilson2,
Lars-Otto Reiersen3
1. Norwegian University of Life Sciences (NMBU), Ås
& University Centre in Svalbard (UNIS), Longyearbyen;
2. Arctic Monitoring and Assessment Programme (AMAP), Tromsø;
3. Arctic Knowledge, Tromsø
The current developments and applications of new, highly sensitive
trace analytical methods allowed identification and quantification of a
still increasing number of contaminants of emerging concern in the
Arctic environment (CEAC = contaminants of emerging Arctic concern).
The recently published and updated AMAP report on CEACs are an
impressive testimony of the wide array of contaminants currently
investigated and monitored in the Arctic Environment.
Earlier source elucidation for legacy organic pollutants identified
long-range transport as a major pollutant source for the Arctic.
However, the thorough investigation of emerging pollutants revealed a
more complex picture. For instance, the evaluation of transport
pathways, chemical properties and fate modelling revealed that
precursor compounds of selected poly- and perfluoralkyl substances
(PFAS) are transported into the Arctic and finally transformed into the
well known the transformation products (i.e. PFOS and PFOA) found
ubiquitously even in Polar regions.
Based on new scientific assessments on compound specific local sources
and complex transport pathways, the Arctic Monitoring and Assessment
Programme (AMAP) has, thus, expanded the current assessment strategies.
A list of more than 300 CEACs is currently discussed for priority
screening in the Arctic.
This list includes modern flame retardants (i.e. phosphorous containing
FRs), personal care products (cyclic siloxanes), pharmaceuticals,
surfactants, food stabilizing chemicals and many more (for
comprehensive information see the current AMAP report on contaminants
of emerging concern).
Our presentation will illustrate the implications of these new findings
for the in-depth environmental research, regional screening, monitoring
activities and regulatory strategies not just for the Arctic
environment. In addition, the final implementation in regional and even
global regulation frameworks will be discussed and elucidated.
The close interdisciplinary linkage between modern environmental
chemistry, toxicology, fate modelling on the one side and monitoring,
environmental assessment and regulation on the other is considered as
mandatory for the balanced pollution regulations in a changing Arctic
with potential conflict scenarios between environmental concerns and
geopolitical, economic and strategic interests in the region.
AN7
Organ on a chip: analysis of mini-organs for personalized
medicine.
Frøydis
Sved Skottvoll
Department of Chemistry, University of Oslo
Current preclinical models (e.g. cell culture- and animal models) often
provide data of poor predictive value, thus complicating and delaying
conclusions on therapeutic interventions. For this reason, recent
advances in tissue engineering and microfabrication have contributed to
the development of an “Organ on a Chip”, a
microfluidic
chip constructed with the purpose of better reconstituting the
complexity of human tissues and organs [1].
As the “Organ on a Chip” platform allows for both
real-time
manipulations and functional readouts, the analytical possibilities are
numerous [2, 3]. Integrating the chip unit with a highly miniaturized
liquid chromatography mass spectrometry system would provide with
unprecedented sensitivity.
Even though the “Organ on a Chip” analytical
platform is
still in its infancy, this microfluidic intervention is predicted to
have a game changing impact on drug screening analysis, diagnosis and
personalized medicine.
References
- Bhatia, S.N. and D.E. Ingber, Microfluidic organs-on-chips.
Nature Biotechnology. Vol. 32 (2014) 760-772.
- Wikswo, J.P., F. Block, D.E. Cliffel, C.R. Goodwin, C.C.
Marasco,
D.A. Markov, D.L. McLean, J.A. McLean, J.R. McKenzie, and R.S.
Reiserer, Engineering challenges for instrumenting and controlling
integrated organ-on-chip systems. IEEE Transactions on Biomedical
Engineering. Vol. 60 (2013) 682-690.
- van Midwoud, P.M., J. Janssen, M.T. Merema, I.A. de Graaf,
G.M.
Groothuis, and E. Verpoorte, On-line HPLC analysis system for
metabolism and inhibition studies in precision-cut liver slices.
Analytical Chemistry. Vol. 83 (2010) 84-91.
AN8
Metabolomics with mass spectrometry: a powerful tool for
clinical analyses.
Skogvold HB1, Sandås EM1,
Østeby A1, Rootwelt H1, Arnesen CE1,
Wilson SRH2, Rønning PO3,
Elgstøen
KBP1
1. Oslo University Hospital, Oslo, Norway
2. University of Oslo
3. Oslo Metropolitan University
Reliable analysis of biomarkers is essential for correct diagnosis and
monitoring of inborn errors of metabolism (IEMs), as is the topic of
this presentation.
We have previously developed an LC-Orbitrap MS method for untargeted
metabolomics of dried blood spots (DBS). This method has been
substantially improved and simplified using only one DBS punch,
extraction with 80% aqueous methanol with formic acid (mix at 700 rpm,
45°C, 45 min). A mobile phase gradient and analysis time of
27.5
min ensures sufficient separation while maintaining good signal
intensity (scan range m/z 50-750, resolution 70 000, electrospray 3.5
kV).
The method is included in research protocols and will be used to detect
differences between healthy controls and patients with various IEMs to
evaluate existing biomarkers and possibly identify new and better ones.
For assessment of the DBS method’s sensitivity in detecting
metabolic changes, we conducted an experiment with controlled diet and
36 hours of fasting in six healthy volunteers.
Analytical evaluation revealed excellent results (retention time
variation 0.2 % and peak area variation 1-5 % for all analytes). The
controlled diet experiment showed that fasting induced changes in the
metabolome as well as clustering of results in Principal Component
Analysis plots from healthy volunteers when changing from a free to a
controlled diet. This demonstrates that the DBS-metabolome is
significantly affected by diet and that the method developed is
suitable to identify metabolic changes.
The DBS metabolomics method showed excellent analytical performance and
ability to identify changes in the blood metabolome reflecting altered
physiologic states induced by dietary intervention. The method will be
used in research to characterize metabolic states and changes in
disease, controlled intervention and during normal daily life
activities in order to identify better biomarkers for diagnosis and
monitoring of patients with IEMs.
AN9
Sporelementer i sjømat og andre marine
prøver -Status og utvikling innen analysemetoder.
Veronika Sele
Havforskningsinstituttet
Sporelementer som arsen, kvikksølv og selen finnes i
spormengder
i miljøet, der sjømat og marine prøver
inneholder
generelt høyere nivå sammenlignet med terrestriske
prøver. For analyser av sporelementer benyttes ofte ICP-MS
(induktivt koblet plasma masse spektrometer). Dette instrumentet er
svært sensitivt og elementspesifikt, og har vokst frem som et
av
de mest anvendte instrumentene innen sporelementanalyser. For noen
sporelementer finnes det ulike kjemiske former, eller element spesier.
Noen spesier, som for eksempel metylkvikksølv og uorganisk
arsen
er mer giftig enn andre spesier, og det er derfor viktig å ha
verktøy for å kunne bestemme disse. For analyser
av
elementspesier blir ICP-MS koblet til en kromatografisk separasjon som
HPLC (høytrykks væskekromatografi) eller GC (gass
kromatografi). Elementer og metaller kan også finnes i form
av
nanopartikler. Analyse av nanopartikler ved bruk av sp-ICP-MS (single
particle ICP-MS) har vokst frem som et forskningsfelt de siste
åra. I denne presentasjonen vil det fortelles om bakgrunnen
for
analyser av sporelementer, og om utviklingen og status innen
analysemetoder for sporelementer; med fokus på analyse av
sjømat og andre marine prøver.
AN10
Measuring with an MC-ICPMS, examples in earth sciences
Cedric Hamelin
Department
of Earth Science, UiB
Multi-Collector
Inductively Coupled Plasma Mass
Spectrometers (MC-ICPMS) are popular research tools in geology, capable
of
measuring heavy radiogenic isotopes (e.g. Nd, Hf, Pb, U-Th) and lighter
stable
isotopes (e.g. Si, Cu, S, Fe), in a variety of material (from sediments
to
igneous rocks). The applications for MC-ICPMS range from geochronology
to
climate change, as well as large-scale geodynamic. The new generation
of these
instruments offers a significant improvement in sensitivity and allow
for
isotopic measurements for reduced sample sizes. In this
presentation, we
will focus on how the Nu Instrument Plasma
2
is used in the Bergen Geoanalytical Facility.
AN11
Den norske NMR-plattformen – En plattform for dine
kjemiske analyser
Jarl Underhaug
Universitetet i Bergen
NMR, eller kjernemagnetisk resonans, er en essensiell teknikk innen
kjemisk-, molekylærbiologisk- og medisinsk analyse. Kjemikere
bruker blant annet NMR til å kvalitetssikre organiske
synteser og
til å karakterisere nye forbindelser, mens
molekylærbiologier bruker NMR til strukturbestemmelse av
proteiner og til å studere interaksjoner med f.eks
legemidler. I
industrien brukes NMR blant annet til kvalitetssikring av
fødevarer. NMR utvikles også til å bli
et
diagnostisk verktøy innen medisin. Dessverre krever NMR
stort og
dyrt utstyr.
Den norske NMR-plattformen er en nasjonal infrastrukturplattform
finansiert av Forskningsrådet. Hovedformålet med
plattformen er å gi forskere, både ved
universitetene og i
industrien, tilgang til moderne høyfelts NMR-spektrometre,
utstyr som ofte er for dyrt for institutter og mindre bedrifter.
Plattformen består av tre moderne, kraftige NMR-spektrometre
som
er plassert ved NTNU, Universitetet i Oslo og Universitetet i Bergen.
Foredraget vil fokusere på NMR generelt, hvilke muligheter
den
nye plattformen gir og hvordan man kan få tilgang til
instrumenteringen. Det blir tatt utgangspunkt i instrumenteringen som
finnes ved UiB, men de fleste analysene kan også
utføres
ved UiO og NTNU.
AN12
Bruk av kjemometriske metoder i analytisk kjemi.
Knut Dyrstad
KD Metrix
The most common multivariate methods used will be shortly presented
followed by examples of applied multivariate analysis in the
development of various analytical methods and corresponding
multivariate / statistical interpretation of analytical output.
Relevant software and how to approach chemometrics for a
‘beginner’ will be discussed.
KA - Katalyse
KAi1
Probing Active Species in Catalysis – Application
of Advanced
X-ray Techniques
Moniek Tromp
Faculty of Science and Engineering, Materials Chemistry —
Zernike Institute for Advanced Materials, University of Groningen, The
Netherlands
moniek.tromp@rug.nl
Detailed information on the structural and electronic properties of a
catalyst or material and how they change during reaction is required to
understand their reaction mechanism and performance. An experimental
technique that can provide structural as well as electronic analysis
and that can be applied in situ/operando and in a time-resolved mode,
is X-ray spectroscopy. Extended X-ray Absorption Fine Structure (EXAFS)
spectroscopy is powerful in determining the local structure of
compounds including amorphous materials and solutions, since long-range
order is not required. Combined X-ray Absorption and X-ray Emission
spectroscopy (XAS and XES resp.) provides detailed insights in the
electronic properties of a material. Detailed information about the
materials in their dynamic chemical active environment can thus be
obtained and structure/electronic – performance relationships
and
reaction mechanisms derived. A combination of spectroscopic techniques
(e.g. UV-Vis, IR) gives complementary information about the system
under investigation.
Over the last years, different approaches have been reported to allow
operando time resolved XAS on catalytic systems, mostly solid-gas. Our
group has developed stopped-flow methodologies allowing simultaneous
time-resolved UV–Vis/XAS experimentation on liquid systems
down to the
millisecond (ms) time resolution [1]. Low X-ray energy systems (light
elements) or for low concentrated systems, longer XAS data acquisition
times in fluorescence detection are required and therefore a stopped
flow freeze-quench procedure has been developed [2]. Pushing the
time-resolution has been achieved by synchronizing the synchrotron
bunches with an optical laser in order to perform fast pump-probe
experiments [3] or micro-reactors for modulation excitation experiments
[4].
Developments in XAS using new instrumentation and data acquisition
methods while selecting specific X-ray energies provide this more
detailed electronic information [5]. High energy resolution XAS, XES
and Resonant Inelastic X-ray Scattering (RIXS) provide very detailed
electronic information on the systems under investigation. The
secondary spectrometer design also opens up lab based spectrometer
designs as will be demonstrated.
The methodologies and instrumentation have been developed and applied
to a wealth of materials science, for homogeneous and heterogeneous
catalysis to batteries and fuel cells as well as art objects. In this
lecture, several examples will be given with an emphasis on homogeneous
catalysis, providing insights in activated species and reaction
mechanisms of selective oligomerisation reaction.
References
- e.g. Tromp M. et al. Organometallics 2010, 29,
3085–3097.
- Bartlett S.A. et al. J. Catal. 2011, 284,
247–258; ACS Catalysis 2014, 4, 4201; Catal. Sci. Techn.
2016, 6, 6237;
Tromp, M. et al, under review.
- Tromp, M. et al. J. Phys. Chem. B 2013, 117(24),
7381–7387.
- Tromp, M. manuscript in preparation.
- e.g. Thomas, R. J. et al. J. Phys. Chem. C 2015, 119(5),
2419–2426; Tromp M. et al, under review.
KAi2
From Homogeneous to Heterogeneous catalysis: Use of
Microporous Solids as Macroligands
Jéróme
Canivet
Univ. Lyon, Univ. Claude Bernard Lyon 1, CNRS, IRCELYON - UMR
5256, Villeurbanne, France.
jerome.canivet@ircelyon.univ-lyon1.fr
At the molecular scale, the integration of the catalytically active
centers into a solid support without loss of performance compared to
the homogeneous analog is still a major challenge. In this context, a
molecularly defined support as macroligand, i.e. a solid acting like
the ligand in the corresponding molecular complex, can be considered as
a key to bridge the gap between molecular and heterogeneous catalysis.
Metal-Organic Frameworks and purely organic microporous polymers are
promising candidates. In particular, porous frameworks made by the
repetition of a coordinating motif, like the bipyridine motif are of a
high interest as far as bipyridines are widely used as chelating ligand
for molecular catalysts.[1,2]. We show that both homogeneous and
heterogenized catalysts follow the same linear correlation between the
electronic effect of the ligand, described by the Hammett parameter,
and the catalytic activity as exemplified in two reactions. This
correlation highlights the crucial impact of the local electronic
environment surrounding the active catalytic center over the long-range
framework structure of the porous support. The gap between molecular
and heterogeneous catalysis has never been so close to being bridged.
This work is carried out within the H-CCAT project that has received
funding from the European Union’s Horizon 2020 research and
innovation
program under grant agreement No 720996. H-CCAT aims at the large scale
production of MOF catalysts and at their use in the industrial
production of pharmaceuticals.
References
- F. M. Wisser, P. Berruyer, L. Cardenas, Y. Mohr, E. A.
Quadrelli, A. Lesage, D. Farrusseng, J. Canivet, ACS Catal., DOI:
10.1021/acscatal.7b03998 (2018)
- F. M. Wisser, Y. Mohr, E. A. Quadrelli, D. Farrusseng, J.
Canivet, ChemCatChem, DOI: 10.1002/cctc.201701836 (2018).
KA1
One-step sol-gel synthesis of Cu/Ordered Mesoporous Alumina
Powders
Ole
Håvik Bjørkedal*, Magnus
Rønning
Department of Chemical Engineering, NTNU, Trondheim
*ole.h.bjorkedal@ntnu.no
Ordered Mesoporous Alumina (OMA) may be synthesized by a sol-gel
process, using micellular polymers as a template for pore structure
[1]. The resulting amorphous alumina powder has narrow pore size
distribution and high surface area, and is well suited for use as a
catalytic support, e.g. by impregnation. An alternative to impregnation
and other similar twostep syntheses is to introduce the metal directly
into the sol-gel process via a suitable precursor.
Inspired by Cu/zeolite systems, Cu/OMA materials have been regarded as
a potential catalyst for NH3-SCR catalyst. A common way of synthesizing
such materials are by two stepmethods such as impregnation of a
support. An alternative procedure (illustrated in Figure 1) is to
synthesize the catalyst in one step, by introducing the metal precursor
directly to the solgel process forming the support structure [2].
One advantage with this procedure is an opportunity to surpass the
limitations of metal loading in ion exchanged zeolitic catalysts while
obtaining a high dispersion of copper sites on the surface. Cu/OMA
materials have also been prepared by incipient wetness impregnation for
comparison. Samples were characterized by N2 physisorption, XRD and
TPR. In situ XAS studies have also been performed to investigate the
materials' behavior under reducing and oxidizing SCR conditions.
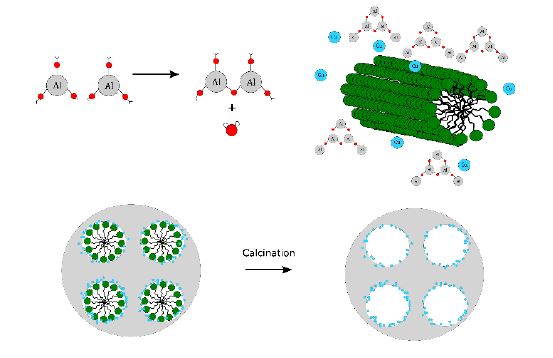
Figure 1: Formation of
Al2O3 (grey) from Al(OPr)3 around
structure-directing micelles forming regular-sized pores. Copper (blue)
is added to the sol.
References
- G. S. Armatas, A. P. Katsoulidis, D. E. Petrakis, and P. J.
Pomonis, “Synthesis and acidic catalytic properties of
ordered mesoporous alumina–tungstophosphoric acid
composites,” J. Mater. Chem., 20, 39, 8631 (2010).
- G. J. B. Voss et al., “Mesostructured alumina as
powders and thin films,” J. Mater. Chem. A, 2, 25,
9727–9735 (2014).
KA2
Methane to methanol conversion over Cu-zeolites –
the XAS view
E.
Borfecchia1*, D. K. Pappas1,
M. Dyballa1, A. Martini2,3,
K. A. Lomachenko4, G. Berlier2,
P. Beato5, C. Lamberti3,6,
S. Bordiga1,2, U. Olsbye1,
S. Svelle1
- Center for
Materials Science and Nanotechnology, Dept. of Chemistry, University of
Oslo, Oslo, Norway
- Dept. of
Chemistry and NIS Centre, University of Turin, Turin, Italy
- The Smart
Materials Research Institute, Rostov-on-Don, Russia
- European
Synchrotron Radiation Facility, Grenoble, France
- Haldor
Topsøe A/S, Kgs. Lyngby, Denmark
- Dept. of
Physics, University of Turin, Turin, Italy
* corresponding author elisa.borfecchia@smn.uio.no
A process allowing energy-effective methane to methanol (MTM)
conversion would represent a major breakthrough for chemical industry.
Cu-exchanged zeolites have been shown to possess Cu active sites able
to cleave the C−H bond of methane at temperatures ≤
200 °C, enabling its stoichiometric transformation into
methanol [1]. The conversion is performed through a stepwise process,
involving high-temperature activation in O2 to generate the active
sites, methane loading at 200 °C, and steam-assisted methanol
extraction.
We have combined laboratory performance testing with in situ/operando
Cu K-edge XAS to establish structure-activity relationships for the MTM
conversion over Cu-CHA [2] and Cu-MOR [3] zeolites. By operando XAS, we
tracked the oxidation state and average coordination of Cu ions during
each step of the process and explored the impact of different
pretreatments and compositional characteristics by in situ XAS.
High-temperature reaction with O2 is evidenced
as a key requirement to form the Cu(II) active sites (Fig. 1a), which
then undergo reversible redox chemistry during the CH4-loading
and CH3OH extraction steps. For Cu-CHA, we
identified a positive linear correlation between the methanol
productivity and the composition-dependent self-reducibility under
high-temperature treatment in He (Fig. 2b), allowing us to rationalize
the composition impact on the productivity for the MTM conversion (Fig.
1c). The most recent research efforts on Cu-MOR have highlighted the
crucial role of keeping consistent conditions for spectroscopy and
performance testing. Having fulfilled this requirement, aided by
high-energy resolution XANES and multivariate analysis [4], we
unambiguously assessed the nuclearity of the Cu-active site in the
investigated series of Cu-MOR materials. These studies highlights the
potential of the combination between XAS and testing at consistent
conditions and paves the way for rationalized material synthesis to
develop an industrial MTM process.
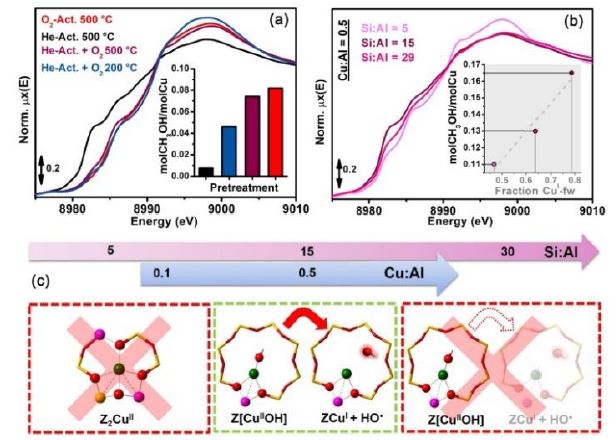 |
Figure
1. In situ XAS of (a) Cu-CHA
with Cu/Al=0.5, Si/Al=12 after different pre-treatments (inset:
corresponding normalized CH3OH productivities); (b) He-activated Cu-CHA
with Cu/Al=0.5 and Si/Al ratio of 5, 15 and 29 (inset: linear
correlation between the normalized productivity and the fraction of
Cu(I) species in the He-activated state. (c) Rationalization of the
effect of Cu-CHA composition on the MTM productivity.
|
References
- E. Borfecchia et al., Chem. Soc. Rev. (2018) in press, doi:
10.1039/c8cs00373d.
- D. K. Pappas et al., J. Am. Chem. Soc., 139 (2017) 14961.
- D. K. Pappas et al., J. Am. Chem. Soc., under review.
- A. Martini et al., Chem. Sci. 8 (2017) 6836.
KA3
Low temperature methanol synthesis catalyzed by Copper
nanoparticles and alkoxide system
Christian
Ahoba-Sam1,2*, Unni Olsbye1
and Klaus-Joachim Jens2
1 - Department of Chemistry, University of Oslo, Oslo,2 -
Department of Process, Energy and Environmental Technology, University
of South-Eastern Norway, Porsgrunn.
* chriaho@kjemi.uio.no
Methanol (MeOH) is identified as a multipurpose molecule, which has a
high potential as a C1 building block for both energy and CO2 storage
[1]. MeOH synthesis at temperatures below 120 oC in a liquid medium
presents the possibility of achieving full syngas conversion per pass
[2]. The Low temperature approach is advantageous over the current
technology for MeOH production since the former is thermodynamically
favourable and gives a high yield per pass. The low temperature
methanol synthesis (LTMS) process involves two main steps, (i) MeOH
carbonylation to form methyl formate and (ii) hydrogenolysis of methyl
formate to form MeOH, illustrated in equations (1) and (2).
𝐶𝑂 + 𝐶𝐻3𝑂𝐻
⇆ 𝐶𝐻3𝑂𝑂𝐶𝐻 (1)
𝐶𝐻3𝑂𝑂𝐶𝐻
+ 2𝐻2 ⇆ 2𝐶𝐻3𝑂𝐻 (2)
Our aim was to characterize, develop and evaluate the LTMS catalyst
system. A once-through catalyst system involving copper (II) salt and
methoxide was used to obtain up to 92 % conversion (> 94 %
selectivity to MeOH) per batch within 2 h at 20 bar syngas pressure and
100 oC temperature. XRD and TEM
characterization of the slurry catalyst system revealed that about 10
± 5 nm Cu nanoparticles were involved in the catalytic
process [3]. Decreasing Cu nanoparticles sizes led to increased MeOH
production due to an increase in active Cu surface area, which enhanced
methyl formate hydrogenolysis. Agglomeration of the Cu nanoparticles in
the course of MeOH production was identified as a major cause for the
deactivation of the Cu nanoparticle component of the LTMS catalyst
system. Furthermore, with the aim of investigating the role of solvents
polarity on the LTMS, MeOH production maximized for solvents with
dielectric constant (ɛ) around 7.2, similar to the polarity of diglyme.
A probe of possible side reactions of the main intermediate revealed
that, in the presence of methoxide, low polar solvents enhanced
decarbonylation of methyl formate while high polar solvents enhanced a
nucleophilic substitution to form dimethyl ether and sodium formate.
References
- Olah, G. A., Angew. Chem. Int. Ed. 44, (2005),
2636–2639
- Christiansen, J. A., (1919), U.S. Patent 1,302,011.
- Ahoba-Sam, C., Olsbye, U., and Jens, K.-J., Catal. Today,
299, (2017), 112-119
KA4
Bimetallic Ni-Fe hydrotalcite-derived catalysts for dry
reforming of methane
Huong Lan Huynh, Henrik Berg, Dori Kalai, Kristian
Stangeland, Zhixin
Yu*
Department of Energy and Petroleum Engineering, University of
Stavanger, 4036 Stavanger, Norway
* corresponding zhixin.yu@uis.no
1. Introduction
Reducing greenhouse gas emissions is the major concern for most of the
world’s leading economies. Amongst proposed technologies, dry
reforming of methane (DRM) has become a promising approach since it
converts natural gas (CH4) and carbon dioxide (CO2)
into syngas (H2 and CO), a valuable building
block for fuels and chemicals. However, the development of active and
stable catalysts for DRM is still challenging. Transition metals (e.g.
Ni, Co) are commonly used due to their good activity but they still
suffer from fast deactivation because of carbon deposition and metal
sintering. Recently, alloying Ni with other metals has attracted much
attention as an alternative catalyst for DRM reaction.
2. Experimental
In this study, a series of bimetallic Ni-Fe catalysts supported on MgAl2O4
were prepared via hydrotalcite (HT) precursors. Ni loading was kept
constant at 20 wt.% while Fe/Ni molar ratio was varied from 0 to 1. Two
different coprecipitation methods were studied, namely conventional
method (low supersaturation) and fast injection method (high
supersaturation) with and without aging step. The characteristics of
catalysts were investigated by X-ray diffraction, nitrogen
adsorption-desorption, hydrogen chemisorption, temperature programmed
reduction and temperature programmed desorption. DRM reaction was
occured at 700 ºC and atmospheric pressure, with
equimolar CH4/CO2 feed at
high gas hourly space velocity.
3. Results and Discussion
Based on XRD data of as-prepared catalysts, the HT-like materials were
synthesized by coprecipitation method without impurities and high
crystallinity. The structure was fully decomposed to oxides after
calcination at 600 ºC. Overall, the catalysts had high BET
surface area and pore volume, representing the advantage of using HT
precursors for catalyst synthesis. The addition of Fe improved the
reducibility and basicity of the catalyst. However, high amount of Fe
content did not favor the Ni dispersion, based on hydrogen
chemisorption data. In DRM reaction, bimetallic catalysts exhibited
better performance than monometallic catalyst. The optimal composition
was found at an Fe/Ni molar ratio of 0.1.
As for catalysts synthesized by fast injection (high supersaturation)
method, Ni and Fe was successfully incorporated in HT-like structure.
The fast-method catalyst (with aging step) had the highest surface
area, pore volume, and narrow pore size distribution, indicating a more
uniformity in particle size. The reducibility and basicity were also
improved. As expected, this catalyst performed the best conversion of
reactants and no deactivation was observed during 18h TOS. Spent
catalysts were studied by XRD; graphite was detected in all samples;
possible Fe3O4 was depicted, supporting the proposed role of Fe to
suppress carbon formation by FeO + C -> Fe + CO reaction [1].
4. Conclusions
High supersaturation synthesis could be an effective approach to
improve the activity and stability of bimetallic Ni-Fe catalysts for
DRM reaction.
References
- S. M. Kim et al., "Cooperativity and Dynamics Increase the
Performance of NiFe Dry Reforming Catalysts," J. American Chem
Soc., 139, 5, 1937-1949
(2017).
KA5
Mesoporous spinel manganese-cobalt oxide catalysts for CO2
hydrogenation to methanol
Kristian
Stangeland1, Dori Yosef Kalai,
Zhixin Yu*2
1 – Department of Energy and Petroleum Engineering,
University of Stavanger, 4036 Stavanger, Norway
* corresponding Zhixin.yu@uis.no
Conventional copper-based catalysts have been extensively studied for
methanol synthesis from CO2, which generally
exhibit insufficient activity and selectivity. Recently, several novel
catalytic systems have been reported to be promising for CO2
hydrogenation to methanol (e.g., Ni(Pd)-Ga, Ni-Sn/InZrO2,
In2O3/ZrO2,
ZnO-ZrO2, and MnOx/Co3O4)
[1-2]. Cobalt-based catalysts are interesting due to its ability to
catalyze different CO2 hydrogenation reactions
to produce methane, methanol, and higher alcohols. It has been shown
that the selectivity of cobalt-based catalysts can be tuned through
optimizing surface properties by utilizing suitable promoters and
supports. Therefore, it will be interesting to identify the nature of
the active site and tuning the selectivity of cobalt-based catalysts
for CO2 hydrogenation. In this work, a series of
mesoporous manganese-cobalt catalysts with different manganese loading
(0, 10, 20, 50, and 100%) were prepared by a modified sol-gel reverse
micelle method. The catalyst were characterized by N2
adorption-desorption, XRD, XPS, ICP-AES, H2-TPR,
CO2-TPD, and TEM. Catalytic testing was carried
out in a continuous flow reactor.
A substantial enhancement in methanol selectivity was observed with the
addition of manganese to mesoporous Co3O4,
which was attributed to a significant enhancement in the surface
basicity. The methanol selectivity was found to strongly depend on the
reduction temperature, and reducing the catalysts at 250 ºC
resulted in the highest methanol selectivity. In contrast, reduction at
200 ºC resulted in an increase in CO selectivity,
whereas methane selectivity increased after reduction at
300 ºC. The highest methanol selectivity and methanol
formation rate was obtained over the 20MnOx-Co3O4
catalyst. The superior performance of 20MnOx-Co3O4
was attributed to enhanced basicity, more easily reducible species,
high surface area, and a higher concentration of well-dispersed
manganese species at the surface. The lower activity of the 50MnOx-Co3O4
catalysts was attributed to manganese blockage of active cobalt sites.
The methane was the majority product over the catalysts regardless of
the reaction conditions. Therefore, further effort is necessary to
increase the methanol selectivity.
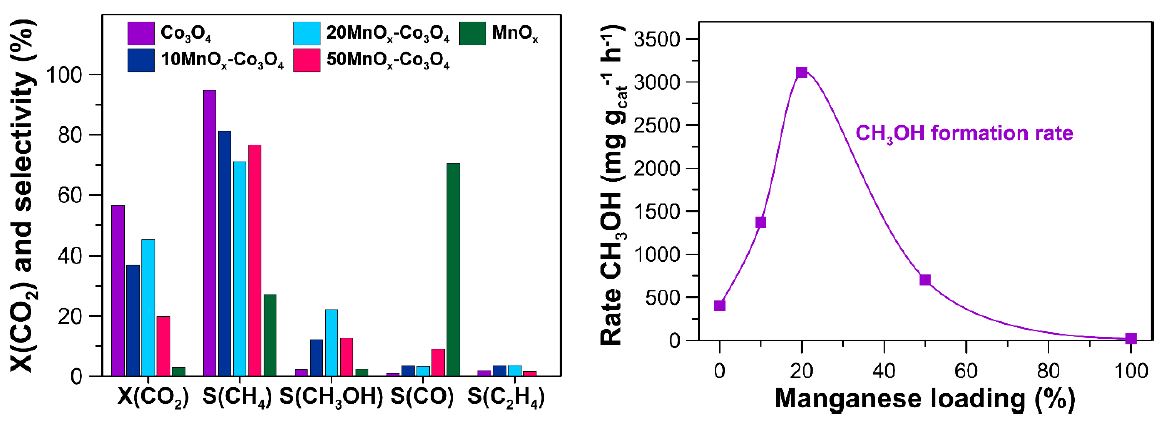
Figure 1. CO2
conversion and product selectivity (left) and the effect of manganese
loading on the methanol formation rate (right) for the Co3O4,
xMnOx-Co3O4
and MnOx catalysts.
References
- Richard AR, Fan M (2017) Low-Pressure Hydrogenation of CO2
to CH3OH Using Ni-In-Al/SiO2
Catalyst Synthesized via a Phyllosilicate Precursor. ACS Catalysis 7 (9):5679-5692
- Li C-S, Melaet G, Ralston WT, An K, Brooks C, Ye Y, Liu
Y-S, Zhu J, Guo J, Alayoglu S (2015) High-performance hybrid oxide
catalyst of manganese and cobalt for low-pressure methanol synthesis.
Nature communications 6:6538
KA6
The CO-induced surface reconstruction on Co(11-20)-a combined
theoretical and experimental investigation
Hilde J. Venvik1*, Marie Døvre
Strømsheim1,
Ingeborg-Helene Svenum2, Mari Helene Farstad1,
Kees-Jan (C.J.) Weststrate3, Anne Borg4
- - Department of Chemical Engineering, NTNU,
7491 Trondheim, Norway
- - SINTEF Industry, 7465 Trondheim, Norway
- - SynCat@DIFFER, Syngaschem BV, P.O. Box 6336,
5600 HH Eindhoven, the Netherlands
- - Department of Physics, NTNU, 7491 Trondheim,
Norway
* hilde.j.venvik@ntnu.no
The surface dynamics of a model Fischer-Tropsch catalyst upon exposure
to CO has been investigated with a combination of experimental and
theoretical methods. The surface of Co(11-20) was chosen as the model
system as it is known to undergo a CO-induced (3x1) surface
reconstruction [1,2] which involves the anisotropic migration of Co [2]
along [0001], initiating from the step edges.
The restructuring was studied with low energy electron diffraction
(LEED), scanning tunnelling microscopy (STM), temperature programmed
desorption (TPD), infrared spectroscopy (IR) and DFT. Three theoretical
model surfaces with a (3x1) periodicity were selected and are displayed
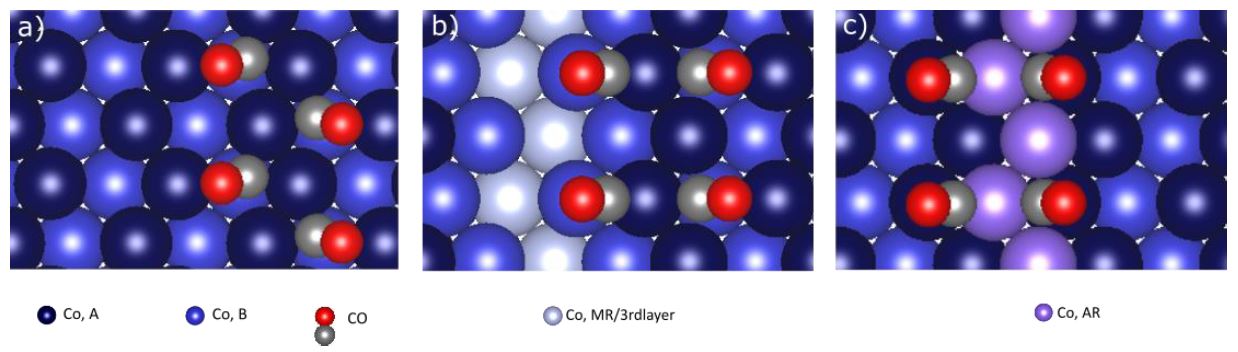
in Figure 1; one unreconstructed surface (a), and two with either a
missing row (MR) (b) or an added row (AR) (c) of Co atoms along [0001],
to represent the
reconstruction. The calculations were performed with the Vienna ab
initio simulations package (VASP) [3].
Figure 1. The calculated
preferred adsorption geometries of 4 CO on (3x2) model surfaces of
Co(11-20) (a) unreconstructed (b) MR and (c) AR.
Calculations showed a slightly higher stability for CO adsorbed on the
reconstructed surfaces than the unreconstructed, with CO in
coordination with the added row yielding the lowest calculated
adsorption energies. The removal of a Co atom from the topmost layer
and its mobility across the surface was investigated through transition
state calculations with climbing image nudged elastic band (CI-NEB)
[4]. These results as well as effects of coverage will be discussed in
relation to the experimental data.
References
- H. Papp, Surf. Sci. 149 (1985) 460.
- H.J. Venvik, A. Borg, C. Berg, Surf. Sci. 397 (1998) 322.
- G. Kresse, J. Hafner, Phys. Rev. B 47 (1993) 558.
- G. Henkelman, B.P. Uberuaga, H. Jónsson, J.
Chem. Phys. 113 (2000) 9901.
KA7
CO2 Hydrogenation over Functionalized
UiO Zr-MOFs
Emil
Sebastian Gutterød, Unni Olsbye*
Centre for Materials Science and Nanotechnology, Department
of Chemistry, University of Oslo, Sem Sælandsvei 26, N-0315,
Oslo, Norway
* corresponding unni.olsbye@kjemi.uio.no
Valorization of CO2 through hydrogenation to products such as CH4,
CO and CH3OH, is attractive for a less
fossil-carbon dependent future [1]. An important research target is
tailored catalytic activity and selectivity by carefully designed
catalyst systems [2]. Metalorganic frameworks (MOFs) are highly tunable
in numerous ways, such as in pore size, in linker functionality and by
metal inclusion. In this work, CO2 hydrogenation
was carried out over Pt-functionalized UiO-67 and UiO-67-binaphtalene
Zr-MOFs at T = 170-280 ºC, p = 1–8 bar, H2/CO2
= 0.2–9 and contact time τ = 0.005–0.04 g
cat min/ml [3]. Under all tested conditions, CO was main product of
reaction at more than 70% selectivity. The selectivity toward minority
products CH4 and CH3OH is
highly dependent on conditions, as well as the MOF characteristics.
Isotope labeling experiments showed that methane is formed from CO2,
via CO. Modification of the MOF with bulky and hydrophobic
binaphtalene-type linkers resulted in an increased CO selectivity and
Eapp of CH4 formation. Comparison to Pt/SiO2
showed very similar activation energy of CO formation, however, the
turnover frequency over Pt/SiO2 was
significantly lower, and no significant formation minority products was
observed. The Pt containing UiO-67 Zr-MOF catalysts showed stable
activity during 60 h of testing.
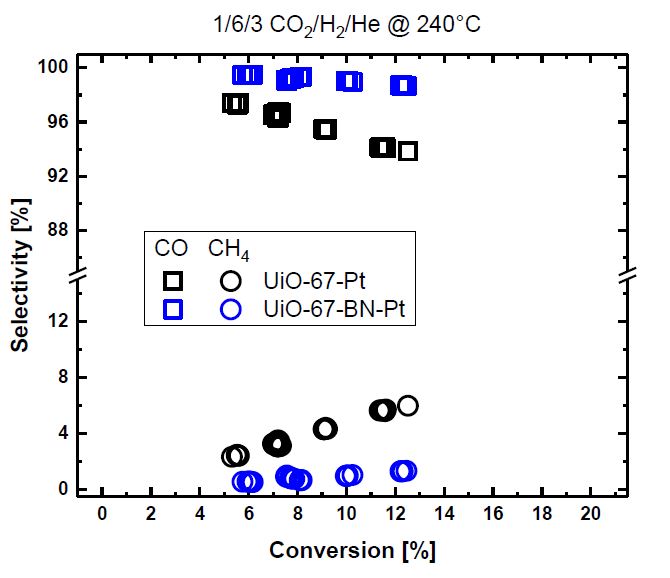
Figure 1. Selectivity of
CO (squares) and CH4 (circles) versus conversion
over UiO-67-Pt (black) and UiO-67-binaphtalene-Pt (blue) during CO2
hydrogenation at ambient pressure, 240 °C, CO2/H2/He
= 1/6/3 and τ = 0.005-0.04 g cat min/ml.
References
- W. Wang, S. Wang, X. Ma, J. Gong, Chem. Soc. Rev., 2011, 40,
3703–3727.
- S. Kattel, P. Liu, J. G. Chen, J. Am. Chem. Soc., 2017, 139,
9739−9754.
- E. S. Gutterød et al., Ind. Eng. Chem. Res., 2017, 66,
13206-13218.
KA8
Operando characterization of Pd-functionalized UiO-67 for CO2
hydrogenation reaction
A.
Lazzarini1,*, G. Kaur1,
E. S. Gutterød1, S. Øien
Ødegaard1, K. P. Lillerud1,
U. Olsbye1, and S. Bordiga1,2
1 – Department
of Chemistry, University of Oslo, Sem Sælands vei 26, 0371
Oslo, Norway
2 – Department of Chemistry, University of Torino, via G.
Quarello 15, 10135 Torino, Italy
*Corresponding author: andrea.lazzarini@smn.uio.no
1. Introduction
Metal-functionalized MOFs are gaining importance in the field of
heterogeneous catalysis [1]. In the recent years, our research group
focused its efforts on the production of Zr-carboxylate based MOFs,
among which, mixed linker UiO-67 materials, represent an interesting
example. 10% of bipyridyl-based linker, substituting the classical bpdc
linker, allowed the grafting of PtCl2 moiety on
the bipyridyl functionality (Figure 1a) [2a]; thermal reduction in H2
results in the formation of metal nanoparticles encapsulated in the MOF
pores [2b]. The obtained material was tested for the CO2 reduction with
H2, showing a good selectivity towards CO and excellent stability at
operat-ing conditions [3]. Among all the metals investigated up to now,
Pd is one of the less explored in the field of MOFs [4], despite its
well-known high activity for hydrogenation and reduction reactions in
general. In the present study, PdCl2-functionalised
UiO-67 was successfully tested in operando conditions for CO2
reduction reaction.
2. Experimental
The sample (hereafter Pd-UiO-67-bpy) was obtained following the
post-synthetic functionalization (PSF) method described in Ref. [3].
PXRD and surface area analysis were performed to check the structural
stability of the material before and after the reaction. SEM microscopy
was used to control the shape of the crystals after each step of the
process. The material was then activated and tested in operando
conditions by means of a combined DRIFT/GC-MS method, able to have a
simultaneous control on both the status of the catalyst and the
products developed during reaction.
3. Results and discussion
Figure 1b shows the PXRD pattern of the sample before and after Pd
impregnation. From that, it is possible to confirm that the MOF
maintained its crystallinity. As expected, the surface area slightly
decreases after the PSF with PdCl2, passing from
2618 m2/g to 2398 m2/g
(Figure 1b - inset). The catalyst was placed inside a Praying Mantis
DRIFT cell for IR measurements and activated in a 10% H2
flow (10 ml/min) at 300°C for 15h, allowing the formation of Pd
NPs inside the pores of the MOF. Maroon curve in Figure 1c shows the
DRIFT spectrum of the sample after activation. All the typical features
of UiO MOF family are present: i) ν(ZrOH) at 3660
cm−1, ii) ν(CH)arom between 3100-3000 cm−1,
iii) ν(COO) between 1550-1300 cm−1, iv)
δ(CH) between 1200-700 cm−1. The MOF is chemically
stable in reaction conditions (240°C, 25% CO2,
75% H2); the only additional IR bands present
are the one at 2350 cm−1 (CO2) and the
one at 2900 cm−1 (probabily due to a slight degradation of
the MOF). The latter however, doesn’t find any degradation
fragment in the acquired chromatograms.
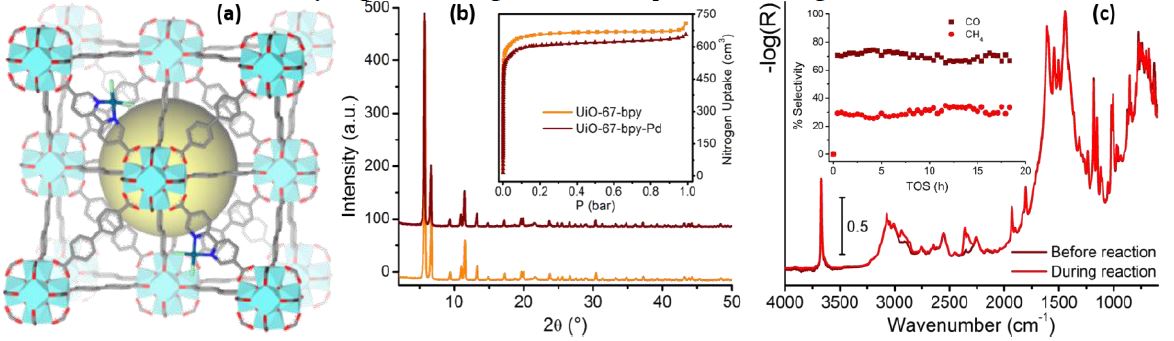
Figure 1. (Part a)
three-dimensional representation of PdCl2-UiO-67-bpy
(adapted from Ref. [2a]). (Part b) PXRD pattern and N2
adsorption (inset) of the UiO-67-bpy MOF before (orange) and after PdCl2
functionalization (maroon). (Part c) DRIFT spectra of H2-activated
Pd-UiO-67-bpy before starting the reaction (maroon) and during CO2
reduction (red). Inset shows the selectivity towards the reaction
products obtained, that are CO (maroon) and CH4
(red).
4. Conclusions
Pd-UiO-67-bpy, shows a slightly lower CO2 conversion compared to the Pt
system. However, the material shows a much higher selectivity towards
CH4 (Figure 1c - inset), making it a good candidate to convert CO2
into CH4.
References
- Y. Han, et al., Chem. Soc. Rev. 43 (2014) 5952; A. Corma,
et al., Chem. Rev. 110 (2010) 4606.
- S. Øien, et al., Chem. Mater. 27 (2015) 1042; L.
Braglia, et al., Faraday Disc. 201 (2017) 265.
- E.S. Gutterød, et al., Ind. Eng. Chem. Res. 56
(2017) 13206.
- H. Fei and S.M. Cohen, Chem. Commun., 50 (2014) 4810; D.
Gao et al., J. Am. Chem Soc., 137 (2015) 4288.
KA9
Adsorption, diffusion, and methylation of light alkenes in
AFI zeotypes: insights from pulse-response TAP measurements
Evgeniy
A. Redekop*, Magnus Morten, Maria Mykland, Unni Olsbye
Centre for Materials Science and Nanotechnology (SMN),
Department of Chemistry, University of Oslo, Norway
* corresponding evgeniyr@smn.uio.no
Physico-chemical interactions of alkenes with the microporous
frameworks of solid acids are crucially involved in determining
outcomes of many industrially-important separation and catalytic
processes such as the Methanol-to-Olefins (MTO) reaction. Herein, the
transient kinetic method of Temporal Analysis of Products (TAP) is
employed to investigate the behavior of C2-C4 alkenes within
metal-substituted aluminophosphates in the limit of zero coverage and
in a broad temperature range (323-673C). At these conditions, the rates
of adsorption, intra-crystalline diffusion, and methylation reaction
are controlled only by the intrinsic properties of alkenes and solid
materials without the interfering influence of pore crowding. Data
suggest that at the temperatures relevant for MTO reactions
(>500K) alkenes have very short residence times within zeotypes,
which, however, are not insignificant for >C2 alkenes and are
increasing with increased acidity of the material. A simple model with
reversible adsorption captures the shapes of the transient responses
well in this temperature range. At lower temperatures, experimental
data cannot be captured by either this simple model or by the model
with spatially resolved intra-crystalline diffusion accompanied by
pseudo-equilibrated adsorption at the pore mouth. We tentatively
attribute the observed behavior to yet unidentified interactions of
alkene double bonds with Brønsted sites of the acidic
zeotypes. The implications of these novel results are discussed in
relevance to precise kinetic characterization of methylation kinetics.
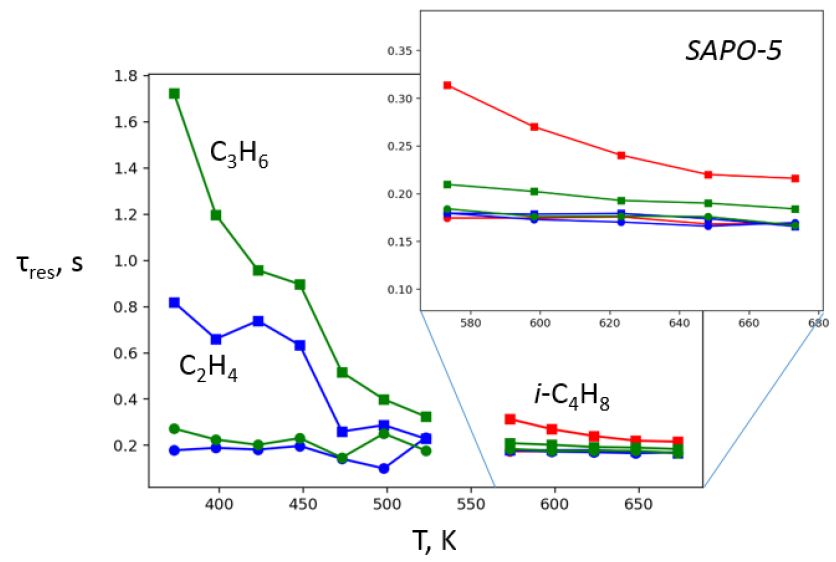
Figure 1. The reactor
residence time of C2-C4 alkenes for SAPO-5, showing that alkenes have
very low delay within the materials in the MTO-relevant temperature
range >600K
References
- U. Olsbye et al., Angew. Chem. Int. Ed. 51 (2012) 5810
– 583.
- K. Morgan et al., Catal. Sci. Technol. 7 (2017) 2416
– 2439
KA10
iCSI – industrial Catalysis Science and Innovation
– a Centre for Research-based Innovation (SFI)
Hilde
J. Venvik*
Department of Chemical Engineering, NTNU –
Norwegian University of Science and Engineering, 7491 Trondheim, Norway
* Hilde.j.Venvik@ntnu.no, https://www.ntnu.edu/icsi
iCSI was appointed Centre for research based innovation (SFI) by the
Research Council of Norway in 2015. iCSI concerns science and
innovation related to industrial processes that are key to Norwegian
land-based industry, global industrial competitiveness, and future
chemical processing and energy conversion with minimum environmental
footprint. These processes supply key sectors of the global market
(catalysts, chemicals, fertilizer, plastics, fuels, etc.); the very
products that impact our food supply and standard of living the most.
iCSI teams the industrial partners Yara, K.A. Rasmussen AS, Dynea,
INOVYN and Haldor Topsøe AS, with the research partners
University of Oslo, SINTEF and Norwegian University of Science and
Technology (NTNU).
The iCSI basic vision is to establish competence and technology that
promotes world-class energy and raw material efficiency for the
industrial partners. iCSI will also be a strong future knowledge base
for the Norwegian chemical industry, and benefit society in terms of
securing jobs, reducing the energy consumption and abating harmful
emissions to the environment. State-of-the-art methodology in
synthesis, characterization, and kinetic investigations are applied to
understand fundamental physical and chemical phenomena critical to the
performance of complex catalysts operating under industrially relevant
conditions. Based on this insight, predictive tools for materials,
chemistry and process optimization can be developed.
The iCSI total budget is MNOK 192 (2015-2023) and NTNU is host
institution. Significant researcher training in the form of ~14PhD and
~6 postdoctoral fellowships is included. iCSI has an extensive
international research interface and a profile of promoting excellence
and leadership of women in research and innovation.
In this contribution, the iCSI Centre Director explains the main
research challenges taken on in iCSI and how the Centre is working on
these. Some research highlights this far will be presented [1-4].
References
- Rout, Kumar Ranjan; Fenes, Endre; Baidoo, Martina
Francisca;
Abdollahi, Reza; Fuglerud, Terje; Chen, De; Highly Active and Stable
CeO2‐Promoted CuCl2/Al2O3 Oxychlorination Catalysts Developed by
Rational Design Using a Rate Diagram of the Catalytic Cycle, ACS
Catalysis, 2016,
6, 7030-7039
- Pappas, Dimitrios; Borfecchia, Elisa; Dyballa, Michael
Martin;
Pankin, Ilia A.; Lomachenko, Kirill A.; Martini, Andrea; Signorile,
Matteo; Teketel, Shewangizaw; Arstad, Bjørnar; Berlier,
Gloria;
Lamberti, Carlo; Bordiga, Silvia; Olsbye, Unni; Lillerud, Karl Petter;
Svelle, Stian; Beato, Pablo, Methane to methanol: structure-activity
relationships for Cu-CHA, Journal of the American Chemical Society, 2017, 139, 42,
14961-14975
- Salman, Ata ul Rauf; Enger, Bjørm Christian;
Auvray,
Xavier; Lødeng, Rune; Menon, Mohan; Waller, David;
Rønning, Magnus, The Catalytic oxidation of NO to NO2 for
nitric
acid production over a Pt/Al2O3 catalyst, Applied Catalysis A, General,
2018,
564, 142-146
- Mom, Rik V.; Ivashenko, Oleksii; Frenken, Joost W.M.;
Groot,
Irene M.N.; Sjåstad, Anja O., The Nucleation, Alloying, and
Stability of Co-Re Bimetallic Nanoparticles on Al2O3/NiAl(110), J.
Phys. Chem. C, 2018,
122, 16, 8967-8975
KA11
Facile synthesis approach for core-shell TiO2–CdS
nanoparticles for enhanced photocatalytic H2
generation from water
Muhammad
Zubair1, Ingeborg-Helene Svenum1,2,
Magnus Rønning1, Jia Yang1*
1 – Department of Chemical Engineering, Norwegian
University
of Science and Technology (NTNU), Sem Sælands vei 4, NO-7491,
Trondheim, Norway
2 – SINTEF Industry, P. O. Box 4760 Torgarden, N-7465,
Trondheim, Norway
* corresponding: jia.yang@ntnu.no
The exponential increase in the burning of fossil fuels to fulfill the
high-energy demands is resulting in severe environmental issues and
depletion of the oil reservoirs in the world. To mitigate the problems
mentioned above, the use of heterogeneous photocatalysts is a promising
way to generate renewable chemical energy in the form of hydrogen (H2)
from water by utilizing solar energy [1]. Among many different
photocatalysts, TiO2
is considered as ideal photocatalysts due to its stability,
cost-effectiveness, and biocompatibility but having a wider band gap
which can absorb only 3-5 % of the solar spectrum. Cadmium sulfide
(CdS) is an ntype photocatalyst, having a band gap of 2.4 eV, which is
a magnificent sensitizer to enhance the light absorption properties of
the wide band gap metal oxide photocatalyst. The major drawbacks of CdS
photocatalysts include a high recombination rate of excited charge
carriers and the photo-corrosion associated with the holes generated in
the valence band of CdS. To address these issues, heterojunction
formation of CdS with other photocatalyst is considered an attractive
approach [2].
The coupling of TiO2 with CdS in the form of
core-shell structure may be the key to get the higher activity and
stability for photocatalytic H2 generation due
to efficient charge separation. Until present, many different TiO2-CdS
catalysts have been investigated for different photocatalytic
applications including water splitting for H2
generation [3], although there is a need to investigate the shell
thickness of TiO2 over CdS and its effect on the
photocatalytic activity of the TiO2-CdS system
by using different hole scavengers.
Herein, we report the synthesis of highly efficient, cost-effective and
stable photocatalyst consisting of TiO2-CdS
core-shell nanoparticles by a facile two-step hydrothermal method. The
crystalline, morphological, optical and band alignment properties of
the developed heterojunction photocatalyst are extensively studies by
respective characterization techniques. The photocatalytic activity of
the TiO2-CdS samples is investigated for the H2
generation
from water under simulated solar light at AM 1.5G conditions by using
various hole scavengers. The most efficient photocatalyst, i.e., TiO2-CdS
(3:2), with the optimized TiO2 shell thickness
over CdS, exhibited the enhanced photocatalytic activity towards H2
generation
from water by producing 954 μmol g-1 h-1 of hydrogen which are
~1.4
and ~1.7 times higher than pure CdS nanoparticles and pure TiO2
respectively. The apparent quantum efficiency of 3.53% was observed by
the atomized sample along with good stability by testing the
photocatalysts for a longer time of consecutive 40 hours. The enhanced
photocatalytic activity and stability of the core-shell TiO2-CdS
nanocomposite is attributed to the
broader solar spectrum absorption, efficient photo-induced charge
separation on the interface of TiO2-CdS due to
the formation of heterojunction and high surface area with a large
fraction of mesopores.
Refrences
- X. Chen, S. Shen, L. Guo, S.S. Mao, Chem. Rev. (2010) 6503.
- M. Reza Gholipour, C.T. Dinh, F. Beland, T.O. Do, Nanoscale
7 (2015) 8187.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y.
Horiuchi, M. Anpo, D.W. Bahnemann, Chem. Rev. 114 (2014) 9919.
KA12
Photocatalytic MOFs for CO2 reduction
Eirik
Mydske Thoresen, Mats Tilset, Mohamed Amedjkouh*
Department of Chemistry, University of Oslo, P.O. Box 1033,
Blindern, 0315 Oslo, Norway
Photocatalysis offer environmentally friendly pathways in a variety of
chemical processes, such as the production of solar fuels. One example
of a solar fuel is methanol formed by photocatalytic reduction of CO2.
Metal-organic frameworks (MOFs) constitute a class of porous and
crystalline hybrid materials that can be functionalized through e.g.
chemical manipulation of the organic linkers.
In this work, new cyclometalated Ru(II) complexes (Figure 1) have been
synthesized1 and incorporated as linkers into the MOF UiO-67 (Figure 2)
by different methods. The resulting functionalized MOFs were
characterized by PXRD, N2 sorption, TGA-DSC,
SEM, EDS, and
UV-Vis spectroscopy. These MOFs show a significantly increased
absorption of visible light compared to MOFs functionalized with
chromophores like amine groups or
non-cyclometalated Ru(II) complexes.
The Ru(II)-functionalized MOFs were tested for photocatalytic CO2
reduction using H2
as reductant in a acetonitrile dispersion. An increasing amount of CO
was detected over time of irradiation of the reaction cell. No CO was
observed in darkness nor when the molecular Ru(II) complex was tested
as catalyst. These results contribute to lay the foundation for the
utilization of CO2 with the help of sunlight.
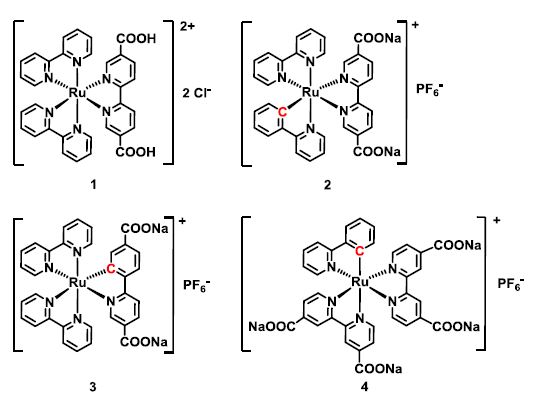
Figure 1. The four Ru(II) complexes that were incorporated into the MOF
UiO-67. |
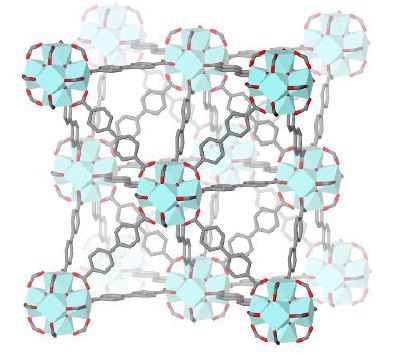
Figure 2. Unit cell
of the MOF UiO-67.
|
Reference
- Thoresen, E. M.; Balcells, D.;
Øien-Ødegaard, S.;
Hylland, K. T.; Tilset, M.; Amedjkouh, M., Cyclometalated ruthenium
complexes with carboxylated ligands from a combined
experimental/computational perspective. Dalton Trans. 2018, 47 (8), 2589-2601.
KA13
Effects of synthesis conditions on the properties and
defectivity of metal-organic framework UiO-67
Gurpreet
Kaur,1 Sigurd
Øien-Ødegaard,1 Knut
Tormodssønn Hylland1, Andrea Lazzarini1,
Sachin Maruti Chavan1, Silvia Bordiga1,
2, Mats Tilset1, Unni Olsbye1
and Karl Petter Lillerud1*
1 – Catalysis
Section, Department of Chemistry, University of Oslo, P.O. Box 1033,
N-0315 Oslo, Norway
2 – Department of Chemistry, NIS and INSTM Reference Centre,
University of Torino, Via G. Quarello 15, 10135 Torino, Italy
*k.p.lillerud@kjemi.uio.no
UiO-67 is a Zr-based MOF with high thermal and chemical stability,
ideally suited for incorporation of additional chemical functionality
using linkers with biphenyl-type geometry. The effect of different
synthesis conditions on the MOF properties is not well understood in
UiO-67 as in UiO-66 [1,2]. Herein, is an improved protocol for the
synthesis of UiO-67 by using a minimal amount of solvent and synthesis
additives, thus diminishing the solvent usage by 82 percent. The
synthesis procedure is then compared with the conventionally reported
method, where we found that more amount of modulator is required as the
amount of solvent is increased for the synthesis of high quality
UiO-67. Moreover, the synthesized compound shows high thermal
stability, and contains considerably less impurities and residues from
the synthesis. This protocol is verified in batch production of up to
170 g MOF, and may thus contribute to significant waste reduction.
Figure 1. Scanning
electron
micrographs of UiO-67 synthesized with increasing amount of modulator
a) 0 eq, b) 3 eq, c) 6 eq, d) 9 eq, e) 12 eq and f) 15 eq in presence
of 50 eq of DMF.
References
- Schaate, A.; Roy, P.; Godt, A.; Lippke, J.; Waltz, F.;
Wiebcke,
M.; Behrens, P., Modulated Synthesis of Zr‐Based
Metal–Organic
Frameworks: From Nano to Single Crystals. Chemistry – A
European
Journal 2011,
17 (24), 6643-6651.
- Gutov, O. V.; Hevia, M. G.; Escudero-Adán, E.
C.; Shafir,
A., Metal–Organic Framework (MOF) Defects under Control:
Insights
into the Missing Linker Sites and Their Implication in the Reactivity
of Zirconium-Based Frameworks. Inorganic Chemistry 2015, 54 (17),
8396-8400.
HI - Kjemiens historie
HI1
Heinrich Goldschmidt: Odd Hassels veileder
Robert Marc
Friedman
Institutt for arkeologi, konservering og historie,
Universitetet i Oslo
Heinrich Goldschmidt (1857-1937) er lite kjent i norsk
vitenskapshistorie. Han flyttet til Norge i 1901 som Peter Waages
etterfølger og gikk av i 1929. På grunn
av mangel
på kilder vet vi lite om hans liv som professor, forsker og
innvandrer utover grunnleggende fakta. Han publisert over to hundre
vitenskapelige artikler og var en dyktig foreleser i nesten tretti
år i Norge. Goldschmidts viktigste bidrag til norsk
vitenskap var sønnen Victor Moritz og studenten Odd
Hassel. Detaljer dukker opp likevel i diverse arkiv som peker
mot
et antall spørsmål som er verdt å
stille, selv om
svarene må vente. Hans vitenskapelige trening og tidlig
profesjonelle erfaring var upåklagelig og nyskapende i at han
kombinert klassisk tysk organisk kjemi med den nye fysikalske kjemien.
Før han kom til Norge arbeidet han i Praha, Zürich,
Amsterdam, Heidelberg. Hvorfor valgte han å forlate
Heidelberg
for Kristiania? Viste han om det store opprøret i Kristiania
mot
å ansette en utlending? Det var bare i siste
øyeblikk at
et flertall i Kollegiet stemte for ansettelsen. Og selv om mange
professorer ønsket ham velkommen, var stemning i
laboratoriet og
kjemimiljøet påvirket av den tidligere motstanden
og av
Goldschmidts fremmede akademisk kultur og fremmed etnisk bakgrunn.
Mange brev viser at rasisme ikke var ukjent i Norge. Muligens var
Goldschmidt og sønnens erfaringer som flinke fremmede ved
Mat-nat fakultetet, et moment hvorfor de alltid sto klar til
å
stille opp for Odd Hassel, som også var betraktet som
annerledes
og en outsider.
HI2
Da
Odd Hassel ble arrestert
Jorunn Sem Fure
Telemark Museum, Skien
Den tyske okkupasjonen fikk store følger for forholdene
på
universitetet. Universitets ledelse ble fra 1941 lagt direkte under
NS-statsråden, og hensikten var å omforme landets
høyeste læresete til en lojal institusjon som
skulle tjene
en nasjonalsosialistisk nyordning av alle samfunnsområder. De
ulike fagene ble utfordret på ulike områder.
Studenter og
ansatte ved Det matematisk- naturvitenskapelige fakultetet ble stilt
over vanskelige valg. Noen fortsatte studier og forskning uavhengig av
de politiske forholdene i den grad det var mulig, noen valgte
å
aktivt støtte den nye ordningen. Blant de som valgte ulike
former for aktiv motstand, finner vi Odd Hassel og flere av hans
kolleger. For disse var det naturlig å sette sine
spesialkunnskaper inn i kampen mot nazismen, men det skjedde ikke uten
risiko og dramatiske følger. Den 15. oktober 1943 ble Odd
Hassel
og ni andre universitetslærer og 63 studenter arrester av det
norske statspolitiet og ført til Berg, det norske
statspolitiets
leir. Maten var elendig og flere ble syke. Hassel ble plassert i
kostebinderiet. Senere ble Hassel overført til Grini og 6.11
1944 ble han løslatt.
HI3
Otto Bastiansen: en karismatisk inspirator
Kari Kveseth
Kjemisk institutt, Universitetet i Oslo
Otto Chr. Bastiansen (1918-1995) var en grunnforskningens
entreprenør. Han engasjerte seg i mye i tillegg
til
forskningen. Han deltok i samfunnsdebatten, i forskningspolitikken og i
utvikling av universitetsdemokratiet. Han var åpen,
uformell, iderik og samarbeidsorientert. Han var en stor
miljøbygger og preget av en aldri sviktende optimisme.
Bastiansen var min faglige veileder, og jeg jobbet som assistent for
ham i flere år. Jeg er engasjert av Kjemisk institutt i et
eget
prosjekt om historien til gassfase elektrondiffraksjon i Norge, et
forskningsfelt startet av Odd Hassel med Bastiansen som viktig
bidragsyter og viderefører. Med utgangspunkt i egen
erfaring, og
historieprosjektet vil jeg i foredraget belyse Bastiansens ulike
engasjementer og vektlegge hans entusiasme, engasjement og evne til
å inspirere.
HI4
Strukturbestemmelsen av benzen – en historie med
mange feilskjær
Leiv K. Sydnes
Kjemisk institutt, Universitetet i Bergen
Faraday sin isolasjon av benzen i 1825 ble innledningen på en
svært aktiv periode i kjemiens historie. Molekylformelen ble
grobunn for mange fantasifull strukturforslag, og mangelen på
kunnskaper om karbons bindingsforhold nørte opp under
kreativiteten. Mange spenstige strukturforslag ble derfor lansert
før Kekulés tanker fikk gjennomslag og ble den
etablerte
sannhet.
I mer enn 50 år bølget diskusjonen fram og tilbake
før konklusjoner ble trukket og feil og mangler ble avdekket
eller sannsynliggjort for godt. Noen vil hevde at en masse tid ble
kastet bort, andre at denne perioden er et glimrende eksempel hvordan
diskusjon skal føres mellom forskere. Uansett har det vist
seg
at flere av de spektakulære forslagene som ble lansert,
senere
har latt seg realisere ved å utføre kjemiske
reaksjoner
på kontrollert vis.
Men historien slutter ikke med det. Etter at Kekulé ble
berømt på grunn av sitt bidrag til
struktur-bestemmelsen
av benzen dukket det opp beskyldninger om det vi i dag kaller
dårlig forskningsetikk. Strukturoppklaringen av benzen er
derfor
beheftet med feil av ymse slag, noe som vil bli presentert i
foredraget.
HI5
Hør Odd Hassel bli intervjuet med mine kommentarer
Bjørn
Pedersen
Kjemisk institutt, Universitetet i Oslo
Per Andersen (1919-2004) og Christian Rømming (1928-2017)
intervjuet professor Odd Hassel (1897-1981) ca. ett år
før
han døde. Både Per og Christian hadde hatt Hassel
som
veileder i hovedfagsstudiet, og begge hadde vært knyttet til
avdeling for fysikalsk kjemi fra de var hovedfagsstudenter så
de
kjente ham godt.
Intervjuet er bevart på en CD som oppbevares på
Kjemisk
institutt. Hassel var ikke enkel å intervjue, og intervjuerne
spurte ham ikke om mye som vi undres over i dag. De spurte ham bl. a.
ikke om hans tid i Berlin 1923-25 da han lærte å
bestemme
krystallstrukturer med røntgendiffraksjon. Hans veileder var
Herman Mark (1895-1992) som senere ble en berømt
polymerkjemiker. I foredraget vil jeg spille av utvalgte deler av CD-en
og gi mine kommentarer.
KI - Kjemometri
KI1
Bruk av kjemometri i medisinske problemstillinger - trenger
vi det?
Tone F. Bathen
MR avbildning og MR spektroskopi er metoder som baseres på de
samme grunnleggende fysiske prinsipper, hvor magnetfelt og
radiobølger gjør det mulig å fremstille
bilder og
spekter. MR avbildning har vært igjennom en rivende utvikling
de
siste 30 årene, og er nå en sentral metode ved
diagnostikk
og behandling av de fleste typer kreft. Samtidig har
bruksområdet
for MR spektroskopi beveget seg fra å være et
verktøy for strukturoppklaring i syntetisk kjemi til
også
å bli et nyttig medisinsk forskningsverktøy for
deteksjon
av metabolske biomarkører (Metabolomikk). Mens kjemometri
har
vært et sentralt verktøy helt fra metabolomikkens
spede
begynnelse, er radiologifaget fortsatt preget av manuell og kvalitativ
lesning.
I denne forelesningen vil jeg benytte eksempler fra MR forskning som
belyser nødvendigheten av kjemometri for å
få full
uttelling av informasjonsrike data.
KI2
Bruk av kjemometri til persontilpasset forebygging av
livsstilssykdommer gjennom livsløpet.
Olav M. Kvalheim
Kjemisk institutt, Universitetet i Bergen
Sykdommer knyttet til livsstil øker som et resultat av en
samfunnsutvikling med blant annet mindre fysisk aktivitet og mindre
variert kosthold med større innslag av hurtigmat og industrielt
fremstilt ferdigmat. Dette påvirker fettstoffskiftet i en
ugunstig retning som kan føre til utvikling av type 2 diabetes
og hjerte/kar-sykdommer. Fettstoffskiftet innebærer transporter
av lipider og kan måles på molekylært nivå i
serum med bruk av kjemiske analysemetoder. Lipidtransporten
utføres av lipoproteiner som kan måles med HPLC og proton
NMR. Ved å lage kalibreringsmodeller mellom HPLC og NMR, kan man
bruke NMR og multivariat modellering har vi utviklet en hurtigmetode
for å kvantifisere opp mot 100 klasser av lipoproteiner, mens det
vanlige lipidpanelet kun gir LDL, HDL og total kolesterol samt total
triglyserid. Mønsteret av lipoproteiner påvirkes av
kjønn og alder i tillegg til livsstilsfaktorer. Forskere ved
UiB, NIH, HVL, Helse Førde og NTNU har deltatt i prosjekter der
tusenvis av serumprøver er analysert og det er laget modeller
som viser hvordan lipoproteinmønsteret påvirkes av
forskjellige faktorer som kjønn, alder, fysisk aktivitet, BMI og
nivå av fettsyrer. Dette åpner muligheten til å
fokusere på forebygging av livsstilssykdom med tidlig
identifikasjon av individer i risikosonen med bruk av
lipoproteinanalyser og påfølgende intervensjon og kontroll
av resultat.
Foredraget vil vise noen eksempler på bruk av kjemometri i denne konteksten.
KI3
Gi prosess-operatørene eierskapet til Big Data.
Harald Martens
Idletechs AS / Inst. teknisk kybernetikk NTNU
Selektivitet: ja, men ikke i rådataene.
Måletekniske rådata kan godt ha store, systematiske
interferens-effekter. Selektivitet sikres i stedet ved hjelp av
multivariat kalibrering basert på f.eks. mange-kanals
hurtigmålinger (spektroskopi osv). Aller best blir slik
kalibrering dersom man kombinerer fysikk-basert modellering og
data-drevet modellering. Dette ble f.eks. brukt da NASA fant vann
på Mars, ved hjelp av spektroskopisk EMSC modellering og PLS
regresjon, implementert i det norske gjør-det-selv
kjemometriprogrammet The Unscrambler.
Industrielle stordata: I
prosessindustrien er digitalisering et brennhett tema. Ved å
sikre en disiplinert strøm av relevante måledata, blir det
i prinsippet mulig å få bedre oversikt over prosessen, og
få bedre alarmhåndtering. Men det vil bare funke dersom man
har gode verktøy til å tolke dataene, og til å
få ned antall falske alarmer.
Ved Inst. teknisk kybernetikk bygger man nå opp et nytt,
industriorientert fagmiljø for å kombinere kjemometriens
multivariate myke modellering med prosess-styringens avanserte
dynamikk-modellering.
I firmaet Idletechs AS – et spin-off fra ITK/NTNU – har vi
kombinert og videreutviklet kjemometri-, kybernetikk- og
maskinlærings- metoder, til å håndtere
«evig-varende», høydimensjonale strømmer av
industrielle eller biomedisinske stordata: «Explainable
AI». Typiske anvendelser: Termisk video og mekanisk
vibrasjonsmåling til overvåking av maskineri, hyperspektral
video av kjemiske prosesser, tørkeprosesser osv.
Mennesket og maskinene:
Metodene våre kan brukes autonomt, uten menneskelig innsats, i
f.eks. satellitter og romfartøyer. Men hovedfokuset er likevel
på å gjøre gode fagarbeidere enda bedre, ved at de
får bedre verktøy til å overblikke og drifte
komplekse produksjonsprosesser. Om kunstig intelligens skaper en
fremtidig kamp mellom menneske og datamaskinene, er vi på parti
med menneskene.
KI4
Kjemometri og magi i matindustrien.
Ingrid
Måge
Nifima
Spektroskopi kan brukes til å måle en lang rekke
kvalitetsegenskaper i mat, både direkte på produksjonslinja
og i laboratoriet. Nærinfrarød (NIR) er den mest brukte
teknikken, men mer høyoppløselige teknikker som for
eksempel Raman og FT-IR har hatt en rivende utvikling de siste
årene. Interessen fra matindustrien er økende fordi denne
type teknologi og metodikk er avgjørende for å løse
noen av dens fremste utfordringer: Å oppnå stabil og riktig
produktkvalitet, optimal utnyttelse av råvarer, minimering av
svinn og dermed en lønnsom og bærekraftig matproduksjon.
Jeg vil vise et knippe eksempler fra forskningsfronten på
spektroskopi og kjemometri i matindustrien. Flere av løsningene
vi har vært med på å utvikle er implementert og
kommersialisert i tett samarbeid med både matindustrien og
utstyrsleverandører. Anvendelsene er så mange og varierte
at det for noen kan framstå som ren magi.
Utvalgte referanser
- Wubshet, S.G., Wold, J.P., Böcker, U., Sanden, K.W., Afseth,
N.K., 2019. Raman spectroscopy for quantification of residual calcium
and total ash in mechanically deboned chicken meat. Food Control 95,
267–273.
- Wubshet, S.G., Wold, J.P., Afseth, N.K., Böcker, U.,
Lindberg, D., Ihunegbo, F.N., Måge, I., 2018. Feed-Forward
Prediction of Product Qualities in Enzymatic Protein Hydrolysis of
Poultry By-products: a Spectroscopic Approach. Food Bioprocess Technol.
- Wold, J.P., Måge, I., Løvland, A., Sanden, K.W.,
Ofstad, R., 2018. Near-infrared spectroscopy detects woody breast
syndrome in chicken fillets by the markers protein content and degree
of water binding. Poult. Sci.
- Måge, I., Wold, J.P., Bjerke, F., Segtnan, V., 2013.
On-line sorting of meat trimmings into targeted fat categories. J. Food
Eng. 115, 306–313.
- Wold, J.P., Kermit, M., Woll, A., 2010. Rapid nondestructive
determination of edible meat content in crabs (Cancer Pagurus) by
Near-Infrared Imaging spectroscopy. Appl. Spectrosc. 64, 691–699.
UN - Kjemiundervisning
UN1
Vitenskap på kjøkkenet - Om kaker som
(ikke) faller sammen og egg som kokes fra innsida og ut
Erik Fooladi
Høgskolen i Volda
Må biffen romtemperes før den stekes? Faller kaka
sammen
hvis du ikke er forsiktig når du tar den ut av ovnen? Og er
det
mulig å lage eplepai helt uten epler? Når du koker
et egg
eller setter en gjærdeig jobber du med kjemiske, biologiske
og
fysiske prosesser på kjøkkenet ditt. Samtidig
bruker du
håndverkskunnskap du har lært av andre eller ved
å
prøve deg fram selv. Men mat er også historie,
kultur,
identitet og sanseerfaringer. Sammen med professor i matvitenskap Anu
Hopia (Universitetet i Turku) har førsteamanuensis Erik
Fooladi
(Høgskulen i Volda) skrevet den
populærvitenskapelige boka
«Kjemi på kjøkkenet: Om hvorfor kaka
faller sammen
og andre kjøkkenhistorier». I boka
ønsker
forfatterne å balansere kjemi, håndverk og
smaksopplevelser, noe som kan gjøre matlagingen mer
spennende,
hodet litt klokere, og maten litt bedre. Og kanskje kan det til og med
fremme kritisk tenkning? I foredraget vil Fooladi presentere boka og
tenkningen bak den, og tilhørerne får
være med
på et sanselig eksperiment.
Bilde
av boka
UN2
Kjemiolympiaden
Hans-Petter
Hersleth og Bjørn Dalhus
Universitetet i Oslo
Kjemiolympiaden har vært arrangert helt siden 1968 og Norge
har
vært med siden 1982. I år er det ca 75 nasjoner som
skal
delta. 4 elever fra hvert land konkurrerer i både praktisk
laboratoriearbeid og teori. Det er "Kjemi-OL komiteen" under NKS som
står for utvelgelse og opplæring av det norske
laget. Dette
gjør vi i tett samarbeid med Kjemisk Institutt og det
matematisk-naturvitenskapelige fakultet ved UiO. Vi står
også for arrangering av den norske kjemi-OL finalen som en
del av
uttakingsarbeidet. I dette foredraget vil vi fortelle litt om hvordan
komiteen arbeider, hvilket opplegg vi har for de norske
uttakskonkurransene, litt om det internasjonale nivået og
også fortelle litt fra den internasjonale finalen.
UN3
Elevforsøk til bruk i kjemi programfag
Karoline
Fægri og Svein Tveit
Universitetet i Oslo
Mange av kompetansemålene i kjemi 1 og kjemi 2 krever at
elevene
gjennomfører elevforsøk. I denne sesjonen
får
deltagerne prøve ut et utvalg elevforsøk som er
knyttet
til kompetansemål i læreplanen for kjemi 1 eller
kjemi 2.
Til hvert forsøk legger vi opp til en fagdidaktisk
diskusjon.
UN4
Triks i Ludo for den digitale kjemilæreren
Asbjørn
Aarflot
Stavanger katedralskole
Sesjonen vil fokusere på bruk av digitale hjelpemidler i
undervisningen og særlig videregående skole. Dette
gjelder
bruk av Marvin sketch til å tegne kjemiske strukturer og
1-HNMR
spekter, autokjeminotasjon i Word med Chemistry formatter og
spesialtegn i Word. En egen del vil være om bruk av Excel til
å laste inn rådata og presentasjon av MS-spekter.
Det blir
lagt opp til egen utprøving med veiledning så ta
med egen
pc eller mac.
KM - Kvantekjemi og
modellering
KM1
Applying coupled cluster theories to solids and surfaces in
the thermodynamic limit
Andreas
Grüneis
Institute for Theoretical Physics, Vienna University of
Technology, Wiedner Hauptstrasse 8-10, 1040 Vienna, Austria
Modern electronic structure theories can predict and simulate a wealth
of phenomena in surface science and solid-state physics.
In order to allow for a direct comparison with experiment, such ab
initio predictions have to be made in the thermodynamic limit,
substantially increasing the computational cost of many-electron
wave-function theories. We present a method that achieves
thermodynamic limit results for periodic solids and surfaces using the
coupled cluster ansatz of quantum chemistry [1].
Computational results for pressure-temperature phase diagrams of carbon
allotropes as well as
adsorption energies of water on hexagonal BN will be presented,
demonstrating the increased efficiency
of our coupled cluster theory implementation for solids and surfaces.
Reference
- T. Gruber, K. Liao, T. Tsatsoulis, F. Hummel and A.
Grüneis , Phys. Rev. X 8, 021043 (2018)
KM2
The Study of Coupled-Cluster Methods Using Strong Monotonicity
Andre
Laestadius, Fabian Faulstich, Simen Kvaal
Hylleraas Centre for Quantum Molecular Sciences, Department
of Chemistry, University of Oslo
By means of an exponential ansatz, the coupled-cluster (CC) method is a
highly successful approach to treat electron correlation. The CC method
and its variations have been analyzed [1,2,3,4] within the ERC project
BIVAQUM [5]. This project studies a generalized variational principle -
the so-called bivariational principle - where the bra and ket of the
Rayleigh-Ritz quotient are treated as truly independent variables. This
brief presentation aims at explaining the basic mathematical concepts
used to prove a locally unique solution of the CC methods. This
includes notions such as strong monotonicity and Lipschitz continuity.
Their connections to a HOMO-LUMO gap and the fluctuation potential
defined as the difference between the system's Hamiltonian and the Fock
operator are here further elaborated.
References
- A. Laestadius and S. Kvaal. Analysis of the
Extended Coupled-Cluster Method in Quantum Chemistry, SIAM J.
Numer. Anal. 56, 660, 2018.
- F. Faulstich et al. Analysis of the
Coupled-Cluster Method Tailored by Tensor-Network States in Quantum
Chemistry, arXiv:1802.05699, 2018.
- A. Laestadius and F. Faulstich. The
Coupled-Cluster Formalism - A Mathematical Perspective,
arXiv:1804.08134, 2018.
- F. Faulstich et al. Numerical and Theoretical
Aspects of the DMRG-TCC Method Exemplified by the Nitrogen Dimer,
to appear on arXiv 2018.
- S. Kvaal, http://www.bivaqum.no
KM3
An efficient pseudo-spectral method for
the description of atomic electronic wave functions -
application to the hydrogen atom in a uniform magnetic field
Clemens Woywod1,
Susmita Roy2, Kiran Maiti3
and Kenneth Ruud4
- Department Chemie, Technische Universitaet Muenchen
- Research Unit of Buhl-Strohmaier Foundation for Cerebral
Palsy and Paediatric Neuroorthopaedics, Orthopaedic Department,
Klinikum rechts der Isar, Technische Universitaet Muenchen
- Max Planck Institut fuer Quantenoptik, Garching
- Hylleraas Centre for Quantum Molecular Sciences,
Department of Chemistry, The University of Tromso - The Arctic
University of Norway
The mapping of an electronic state on a real-space support lattice may
offer advantages over a basis set ansatz in cases where there are
linear dependences
due to basis set overcompleteness or when strong internal or external
fields are present.
Such discretization methods are also of interest because they allow
for the convenient numerical integration of matrix elements of local
operators.
We have developed a pseudo-spectral approach to the numerical solution
of the time-dependent
and time-independent Schroedinger equations describing electronic
motion in atoms and atomic ions
in terms of a spherical coordinate system.
A key feature of this scheme is the construction of a Variational Basis
Representation (VBR) for the non-local component
and of a Generalized Finite Basis Representation (GFBR) for the local
component of the
electronic Hamiltonian operator.
Radial Hamiltonian eigenfunctions χnl;β(r)
of the H atom-like system and
spherical harmonics form the basis set.
Two special cases of this approach are explored: In one case, the
functions of the field-free H atom
are used as the elements of the basis set, and in the second case, each
radial basis function has been obtained by variationally optimizing
a shielding parameter beta to yield a minimum energy for a particular
eigenstate of the H atom
in a uniform magnetic field.
We derive a new quadrature rule of nearly Gaussian accuracy for the
computation
of matrix elements of local operators between radial basis functions
and
perform numerical evaluation of local operator matrix elements
involving spherical harmonics.
With this combination of radial and angular quadrature prescriptions we
ensure to a good approximation the discrete orthogonality of
Hamiltonian eigenfunctions
of H atom-like systems for summation over the grid points.
The pseudo-spectral approach presented here is applied to two model
systems: the field-free H atom and
the H atom in a uniform magnetic field. The results demonstrate the
potential of this method for the description of challenging systems
such as highly charged atomic ions.
Reference
- Woywod, Roy, Maiti and Ruud, Chemical Physics, submitted
KM4
Modelling nanoscale friction of adsorbed molecules
Astrid
de Wijn
Department of Mechanical and Industrial Engineering,
Norwegian University of Science and Technology, 7491 Trondheim, Norway
Friction between solid surfaces is an important phenomenon in everyday
life. A large portion of the total energy production in industrialised
countries is lost through friction and wear. Friction is a
very
complex phenomenon with dynamics happening on many length and time
scales. At the most basic level, however, is the dissipation
at
the nano-scale level. At this level, real sliding interfaces
can
still be fairly complex. Often, there are molecules adsorbed
on
the surfaces, originating from the atmosphere or additives that have
been put in a lubricant to protect the surface from wear, corrosion,
etc.
I will discuss theoretical approaches to studying how adsorbed
molecules affect the friction at the nano scale, using simple models
and molecular-dynamics simulations.
KM5
Molecular dynamics in a density dependent inhomogeneous
dielectric
Sigbjørn
Løland Bore, Hima Bindu Kolli, Toshihiro
Kawakatsu, Giuseppe Milano, and Michele Cascella
University of Oslo
A new molecular dynamics method for
computing electrostatic forces in a inhomogeneous density dependent
dielectric is presented. Using a hybrid particle field approach[1],
forces acting on charged particles and the particles making up the
dielectric are derived. These forces are computed from the
electrostatic potential, obtained by solving the generalized
Poisson's equation. Benchmarking of the method, shows that it is able
to describe partitioning phenomena due to differences in dielectric
properties in a binary phase system. Furthermore, for a charged lipid
bilayer, it shown that forces on the particles making up the
dielectric have a significant contribution to the force density around
the membrane, with net effect of contracting the width of the
membrane.
References
- Hybrid particle-field molecular dynamics
simulations for dense polymer systems
Giuseppe Milano1, and Toshihiro Kawakatsu
The Journal of Chemical Physics, (2009) 130:21
KM6
Self-Assembly of α-Tocopherol Transfer Protein
Nanoparticles: A Patchy Protein Model
Raphael Peltzer,† Hima Bindu Kolli,†
Achim Stocker,‡ and Michele Cascella,†
†Department of Chemistry, and Hylleraas Centre for
Quantum Molecular Sciences, University of Oslo, P.O. Box 1033,
Blindern, 0315 Oslo, Norway
‡Department of Chemistry and Biochemistry, University of
Bern, Freiestrasse 3, 3012 Bern, Switzerland
I describe the mechanism of self-aggregation of α
–tocopherol transfer protein into a spherical
nanocage employing Monte Carlo simulations. The protein is modeled by a
patchy coarse-grained representation, where the
protein−protein interfaces, determined in the past by X-ray
diff raction, are represented by simplified two-body interaction
potentials. Our results show that the oligomerization kinetics proceeds
in two steps, with the formation of metastable trimeric units and the
subsequent assembly into the spherical aggregates. Data are in
agreement with experimental observations regarding the prevalence of
different aggregation states at specific ambient conditions. Finally,
our results indicate a route for the experimental stabilization of the
trimer, crucial for the understanding of the physiological role of such
aggregates in vitamin E body trafficking.
KM7
DrawMol a new program to visualize molecular properties -
Application to Magnetically Induced Current of Helicene molecules
Vincent
LIEGEOIS
Laboratory of Theoretical Chemistry, Namur Institute of
Structured Matter (NISM), University of Namur, rue de Bruxelles 61,
5000 Namur, Belgium.
DrawMol is a full-featured program to build molecular structures from
scratch (and to generate the input files for Gamess-US and Gaussian
quantum chemistry packages) as well as to visualize molecular
properties. In addition to the visualization of the structures, the
molecular orbitals and the dipole moments that are commonly found in
other programs, DrawMol also represents the polarizability and
hyper-polarizability using the unit sphere representation, the
polarizability ellipsoid, the vibrational normal modes together with
the IR vectors, the NMR chemical shifts, and the magnetically induced
current density. This program is for Mac only and is available on sale
on the Mac App Store since November 2016.
In this contribution, I will present some magnetically induced current
density calculations performed on helicene molecules.
For that, Gaussian 16 program together with the GIMIC code [1] have
been used. Starting from [6]Helicene molecule, we have investigated
larger and larger helicenes up to 20 fused benzene rings. Among the
interesting features, we have noticed that the two extrema rings bear
the largest induced current values as well as the smallest NICS(0)
values. From [7]Helicene, we have substituted 1, 3 and 4 benzene rings
by either pyrrole or thiophene moieties. Results have shown that the
5-membered rings were having the smallest current value.
Fig. 1. Streamline representation of the magnetically induced current
of [7]helicene. The external magnetic field is pointing toward the
reader.
Reference
- M. Rauhalahti, S. Taubert, D. Sundholm, V.
Liégeois, Phys. Chem. Chem. Phys. 19 (2017) 7124.
KM8
Relativistic real-time and linear response TDDFT approaches
to electron absorption and circular
dichroism spectroscopies
Lukas
Konecny1, Marius Kadek1,
Stanislav Komorovsky2, Kenneth Ruud1,
Michal Repisky1
1Hylleraas Centre for Quantum
Molecular Sciences, UiT—The Arctic university of Norway,
Tromsø, Norway
2Institute of Inorganic Chemistry, Slovak
Academy of Sciences, Bratislava, Slovakia
We present implementation and applications of two methods for the
evaluation of electron absorption (EAS) and circular dichroism
(ECD) spectra available in the relativistic quantum chemistry density
functional theory (DFT) program ReSpect
(www.respectprogram.org). First is the electron dynamics (real-time
time-dependent DFT) based on a direct propagation of electron
density matrix in time. The spectra are obtained by the Fourier
transformation of time-dependent induced electric or magnetic dipole
moments recorded during the propagation. Such a method allows to access
spectra in various regions, including near-resonant
frequencies, from a single simulation, as well as to treat molecules
subjected to strong or arbitrarily time-dependent external
fields without the need to calculate the response kernels. [1]
A cost-efficient alternative in the weak field regime is the damped
linear response theory that is based on perturbation expansion.
An algebraic response equation is solved using the iterative subspace
algorithm in the frequency domain to directly yield the
spectral function for the frequencies of interest. [2]
Both scalar relativistic effects and spin-orbit coupling are treated
variationally by means of the 4-component Dirac–Coulomb
Hamiltonian represented in the basis of restricted kinetically balanced
Gaussian functions exploiting the noncollinear Kramers
unrestricted formalism, as well as by the computationally efficient
quasi-relativistic 2-component X2C Hamiltonian obtained from the
original 4-component Hamiltonian by adiabatic decoupling transformation
formulated entirely in matrix algebra.[3].
We demonstrate the performance of the developed methods by calculating
EAS and ECD spectra of a number of benchmark systems
including heavy-element containing molecules in valence as well as
X-ray regions.
References
- M. Repisky, L. Konecny, M. Kadek, S. Komorovsky, O. L.
Malkin, V. G Malkin, K. Ruud, J. Chem. Theory Comput.,
11, 980 (2015).
- L. Konecny, M. Repisky, K. Ruud, S. Komorovsky, in
preparation
- L. Konecny, M. Kadek, S. Komorovsky, O. L. Malkin, K. Ruud,
M. Repisky, J. Chem. Theory Comput., 12,
5823 (2016).
KM9
Time-Dependent Coupled-Cluster Theory
Thomas Bondo Pedersen and Simen Kvaal
Hylleraas Centre for Quantum Molecular Sciences, Department of Chemistry, University of Oslo
Coupled-cluster theory is the most successful wave function-based method in quantum chemistry today.
It provides highly accurate energies and properties of electronic ground states, excitation energies and excitation
strengths, and excited-state
properties, as long as the ground state is dominated by a single electron configuration.
Spectroscopic properties are computed using perturbation theory with the underlying assumption that the
external field is sufficiently weak. Motivated by the investments over the last decade in the development of
extremely brilliant and coherent laser sources, we present a nonperturbative approach to spectroscopic
processes based on direct propagation of the coupled-cluster state in the presence of an intense and short laser
pulse. The main focus of the talk will be on the time-propagation itself, interpretation of the time signals,
and on the suitability of coupled-cluster theory for the description of molecular electronic systems in the
extreme environment of a laser pulse.
KM10
Selective CO2 conversion
with Rh/bisphosphine-thiourea
(ZhaoPhos)
catalyst
Ljiljana
Pavlović,
Janakiram Vaitla, Annette Bayer, Kathrin H. Hopmann
Hylleraas
Centre for Quantum Molecular Sciences,
Department of Chemistry, UiT The Arctic University of Norway ljiljana.pavlovic@uit.no
Transition
metal-catalzyed enantioselective hydrocarboxyalation of
α,β-unsaturated esters could provide a method to
synthesise chiral
carboxylic acids, which are the main components of many drugs. Thus,
the design
of enantioselective and sustainable catalysts for this reaction with CO2,
presents a big challenge in the pharmaceutical industry. Only one
asymmetric CO2-based
hydrocarboxylation reaction has been reported, involving a rhodium
complex with
a bidentate SegPhos ligand, which in the presence of ZnEt2
carboxylates α,β-unsaturated esters with up to 66%
enantiomeric
excess. [1] Our previous theoretical study revealed that during during
the C -CO2
bond formation, the CO2
molecule interacts with neither the rhodium complex nor the
organozinc
additive. [2] This
appears to be in
contrast to other CO2 insertion reactions, where
CO2−metal
interactions have been predicted. Additinally, the substrates show an
unusual
coordination mode during CO2
insertion, with the nucleophilic carbon
positioned up to 3.6 Å away from rhodium.
Based on these findings, we further investigated the potential for
enantioselective CO2 conversion. The asymmetric CO2-based
hydrocarboxylation reaction has been studied using density functional
theory (PBE-D2/IEFPCM). For this purpose, chiral transition metal based
catalysts can be employed, which through selective interactions with
the substrate, are able to favour the formation of only one
enantiomer. Many ligands were studied: PHOX, SegPhos and ZhaoPhos, but
all of them gave little selectivity. On the contrary, the use of a
novel and modified ZhaoPhos ligand, that has not been previously used
in hydrocarboxylation, gave a good selectivity in
computations.
It has been known that thiourea, which is linked to the ligand, works
as a hydrogen donor, thus it activates the substrate and
provides
high conversion and excellent enantioselectivity in many Rh-based
hydrogenation reactions.[3]Futhrer investigation of this efficent
catalyst are ongoing in our labaratory.
References
- Kawashima, S.; Aikawa, K.; Mikami, K.; Eur. J.
Org. Chem.
2016, 3166-3170.
- Pavlovic,
Lj.; Vaitla, J., Bayer, A.;Hopmann, K.H.; Organometallics 2018 37,
941−948
- Q.
Zhao, S. Li, K. Huang, R. Wang and X. Zhang; Org.
Lett., 2013,15,
4014–4017
KM11
Decarbonylative dehydration of fatty acids: New
mechanistic insight
Sondre
H.H. Eliasson, Anamitra Chatterjee, Vidar R. Jensen
Kjemisk Institutt, Universitetet i Bergen
Linear α-olefins(LAOs) are key commodity chemicals and
petrochemical intermediates currently produced from fossil resources.
However, renewable resources may also provide α-olefins. As
we have recently reviewed,[1] one attractive biomass for such
production is fatty acids and their derivatives. From fatty acids,
α-olefins may be reached via transition-metal-catalyzed
deoxygenation, the perhaps most promising variation of which is
decarbonylative dehydration.[1] However, the best decarbonylative
dehydration catalysts obtained to date are not active and stable enough
for industrial use. Fortunately, designing more active and stable
catalysts is now being facilitated by the arrival of the first
mechanistic insight from density functional theory (DFT) studies[2-5]
and the first well-defined precatalyst for this reaction,
Pd(cinnamyl)Cl(DPEPhos),[6] which offers superior activity at
relatively low temperatures (110 °C). Recent DFT calculations
show how the DPEPhos ligand contributes to a low overall barrier and
high α-selectivity by switching from bidentate to monodentate
binding mode prior to the rate-determining β-H-elimination
step.[7] Retaining one of the Pd–P bonds
of the DPEPhos ligand while dissociating acetate and CO is advantageous
in dipolar solvents such as DMPU or the green alternative
γ-valerolactone (GVL),[8] and eliminates the large excess of
phosphine otherwise needed for catalyst regeneration and sustained
activity.[1] The effect of the DPEPhos binding mode switching suggests
that new and improved ligands might be designed that combine strongly
coordinating with more labile binding sites.
Figure 1.
References
- A. Chatterjee, S. H. Hopen Eliasson and V. R. Jensen,
Catalysis Science & Technology, 2018, 8, 1487-1499.
- M. A. Ortuño, B. Dereli and C. J.
Cramer, Inorg. Chem., 2016, 55, 4124-4131
- A. John, M. O. Miranda, K. Ding, B. Dereli, M. A.
Ortuño, A. M. LaPointe, G. W. Coates, C. J.
Cramer and W. B. Tolman, Organometallics, 2016, 35, 2391-2400
- A. John, B. Dereli, M. A. Ortuño, H.
E. Johnson, M. A. Hillmyer, C. J. Cramer and W. B. Tolman,
Organometallics, 2017, 36, 2956-2964.
- S. H. H. Eliasson, A. Chatterjee, G. Occhipinti and V. R.
Jensen, Inorganics, 2017, 5, 87.
- A. Chatterjee, S. H. H. Eliasson, K. W.
Törnroos and V. R. Jensen, ACS Catal., 2016, 6,
7784-7789.
- S. H. H. Eliasson, A. Chatterjee and V. R. Jensen, The
mechanism for selective and active decarbonylative dehydration of fatty
acids, manuscript in progress
- A. Chatterjee, S. H. H. Eliasson and V. R. Jensen, Green
solvent for the synthesis of linear a-olefins from fatty acids,
unpublished work.
KM12
Catalytic
Hydrogenation of Amides to Methanol and Amines from a Computational
Perspective.
Lluis Artus Suarez, David Balcells, Mats Tilset, Ainara Nova
University of Oslo
CO2 is abundant, cheap, non-flammable and has low toxicity, making it
an ideal renewable carbon feedstock. Recently, the conversion of CO2 to
methanol was performed in a one-pot reaction with a ruthenium
bifunctional catalyst, in the presence of amines.[1] The participation
of amides as intermediates in the mechanism of this reaction prompted
us to study their reduction to methanol, with the aim of developing a
rational approach to the design of more active and robust catalytic
systems. In this work, the mechanism for the hydrogenation of
formanilide and dimethyl formamide (DMF) to methanol with an iron
catalyst (Figure) has been studied with a DFT method and compared to
the experimental results of Bernskoetter and Hazari.[2] The
microkinetic models derived from the DFT calculations reproduced the
high conversions obtained with formanilide and the need of using the
latter as co-catalyst in the hydrogenation of DMF. The computational
studies revealed a complex reaction network arising from three
consecutive processes; namely 1) the hydrogenation of the amide C=O
bond, 2) the protonolysis of the C–N bond of an hemiaminal
intermediate and 3) the hydrogenation of formaldehyde. Interestingly,
the mechanism of process 2) depends on the nature of the substrate.
Figure. Reaction mechanism postulated for the iron-catalyzed
hydrogenation of amides.
References
- M. S. Sandford et. al., J. Am. Chem. Soc. 2015, 137,
1028-1031.
- N. Hazari, W. H. Bernskoetter et. al., Organometallics,
2017, 36, 409-416.
KM13
DigiBiotics: discovering novel antimicrobial molecules from
Arctic marine fungi
Karolina
Solheimslid Eikås, Kenneth Ruud, Maarten
Beerepoot and Bjørn Olav Bransdal
Hylleraas Centre for Quantum Molecular Sciences, Department
of Chemistry, UiT The Arctic University of Norway
The DigiBiotics project aims to discover novel antimicrobial molecules
from Arctic marine fungi. My part of the project is to develop a novel
computational protocol for structure determination of the complex and
chiral cyclic polypeptides found in the fungi. The molecules of main
interest in this project are larger than the clinical antibiotics used
today and there is a need for an efficient and reliable protocol for
structural determination of these molecules. Due to the large and
complex molecule structure, this protocol will combine experimental and
computational spectroscopy to reach the gold standard for the
determination of absolute configuration. Several different
spectroscopies will be used, but my project will focus on the
vibrational chiroptical methods Raman Optical Activity (ROA) and
Vibrational Circular Dichroism (VCD). In my presentation I will point
out why we need these types of spectroscopy and the strategy to develop
this protocol.
Figure 1. From Marine Fungi to commercial antibiotics. The picture to the
left is taken by Marte Jensen (UiT Norges arktiske universitett), the
figure in the middle is taken from ref [
1] and to the right an
illustration:
www.colourbox.com
Reference
- Kathrin H. Hopmann; Kenneth Ruud; Magdalena Pecul; Andrzej
Kudelski; Martin
DraÄÃnský; Petr
BouÅ™; J. Phys. Chem. B 2011, 115, 4128-4137. DOI: 10.1021/jp110662w
KM14
Peptide fibrillization revealed by two-photon absorption
Maarten Beerepoot1,
Md. Mehboob Alam1, Kenneth Ruud1,
Robert Zalesny2 and Piotr Hanczyc3
1Hylleraas
Centre for Quantum Molecular Sciences, Department of
Chemistry, UiT The Arctic University of Norway,
maarten.beerepoot@uit.no
2Department of Physical and Quantum Chemistry,
Faculty of Chemistry, Wroclaw University of Science and Technology,
Poland
3Institute of Physical Chemistry, Polish Academy
of Sciences, Warsaw, Poland
Peptide fibrillization and the resulting amyloid fibres are
responsibles for various diseases among which Parkonson's and
Alzheimer's.
Experimental work has shown strong non-linear absorption as a result of
peptide fibrillization, which has been explained by though-space
intermolecular interactions between aromatic residues in different
peptides in the same fibril [1].
In another study, quantum chemical calculation have shown that a
similar mechanism of through-space charge transfer is responsible for
enhanced two-photon absorption in the globular yellow fluorescent
protein [2].
The aim of the present project is to use experimental
multiphoton absorption techniques in combination with quantum chemical
calculations to investigate whether intermolecular charge transfer can
indeed explain the enhanced multiphoton absorption in protein fibrils.
The combination of experimental evidence with mechanistic insights from
quantum chemical calculations may help in the development of new
techniques to detect amyloid fibrils and/or in the development of new
materials for applications in nanotechnology.
References
- Hanczyc, Samoc and Norden, "Multiphoton absorption in amyloid protein
fibres", Nature Photonics 7 (2013),
p. 969.
- Beerepoot, Friese and Ruud, "Intermolecular charge transfer enhances two-
photon absorption in yellow fluorescent protein", Phys.
Chem. Chem. Phys. 16 (2014), p. 5958
KM15
Molecular vibrations with polarizable embedding
Karen
Oda Hjorth Dundas, Kenneth Ruud, Maarten Beerepoot, Magnus
Ringholm, Magnus Olsen
Hylleraas Centre for Quantum Molecular Sciences, Department
of Chemistry, UiT The Arctic University of Norway
Methods combining quantum mechanical (QM) methods and molecular
mechanics (MM) methods are important tools when it comes to studying
larger molecular systems. One example of such a QM/MM method is
Polarizable Embedding (PE) where a central core is modelled with a QM
method of choice (DFT) and the surrounding environment is classically
modelled by placing multipoles an polarizabilities on each atomic site.
In this way you can in addition to looking at the QM region calculated
it's electrostatic and induced interaction with the environment. We are
now working on combining this with a recursive response theory library
so that we can look at various molecular properties, and specifically
molecular vibrations, for large molecular systems. This will allow us
to look at for instance IR or Raman properties for solutions or
biomolecules in an efficient fashion.
KM16
RNA structure and dynamics combining molecular simulations
and experiments.
Giovanni Bussi
SISSA, Trieste, Italia
RNA structure and dynamics combining molecular simulations and
experiments
.
Ribonucleic acid (RNA) has a fundamental role in cell biology. However,
experimental characterization of RNAs dynamical behavior at atomistic
level is difficult. Molecular simulations (MD) at atomistic detail in
combination with state-of-the-art free-energy techniques could in
principle bridge the gap providing an unparalleled perspective on the
mechanism and dynamics of RNA folding and conformational transitions.
However, current empirical force fields used to model RNA are not yet
accurate enough to predict structural dynamics in agreement with
solution phase experiments. In this talk I will show how it is possible
to combine solution experiments and MD simulations to reliably predict
RNA structural dynamics using both the maximum entropy and the maximum
parsimony principles [1,2] in order to recover excited states that are
difficult to directly visualize in experiment. In addition, I will show
how experimental data can be used in order to systematically improve
the accuracy of existing force fields [3].
[1] Cesari, Reisser, Bussi, Computation (2018).
[2] Reisser et al, in preparation (2018).
[3] Cesari et al, in preparation (2018).
KM17
Role of time-reversal symmetry in relativistic band-structure
calculations of solids
Marius
Kadek, Michal Repisky, Kenneth Ruud
Hylleraas Centre for Quantum Molecular Sciences, Department
of Chemistry, UiT – The Arctic University of Norway,
Tromsø, Norway
In the talk, I will outline the concept of the time-reversal symmetry
in the
context of relativistic electronic structure theory of periodic
systems. I will
explore the consequences of the time-reversal symmetry and the
spin–orbit
coupling on the structure of operators and matrices expressed using the
restricted kinetically balanced Gaussian-type orbitals [1], and extend
the
quaternion formalism [2,3] to reciprocal space [4]. Some properties of
spin–orbit-coupled two-dimensional materials will be
discussed, as well
as possible future directions and applications of our relativistic
Gaussian-based all-electron method that employs the 4-component
Dirac–Coulomb Hamiltonian.
Fig. 1. Dirac cones
appearing in the band structure surface plot of the highest occupied
band (blue) and the lowest unoccupied band (orange) of the
two-dimensional germanene.
References
- R. E. Stanton and S. Havriliak, J. Chem. Phys. 81, 1910
(1984).
- T. Saue, K. Fægri, T. Helgaker, and
O. Gropen, Mol. Phys. 91, 937 (1997).
- L. Konecny, M. Kadek, S. Komorovsky, K. Ruud, and M.
Repisky, J. Chem. Phys. (2018), (to be published).
- M. Kadek, M. Repisky, and K. Ruud. in preparation.
KM18
Contribution of the aromatic amino acids in membrane binding
of peripheral proteins using Free Energy Perturbation.
Qaiser
Waheed, Hanif M. Khan and Nathalie Reuter 1
Department of biological sceinces and Computational biology
unit, University of Bergen
1. Department of Chemistry and Computational biology unit, University
of Bergen
Membrane binding of peripheral proteins is governed by
contributions from the partitioning of individual amino acids and from
specific interactions with the lipids. Dissecting these individual
amino acid contributions is challenging from equilibrium molecular
dynamics simulations. On the other hand, methods like free energy of
perturbation (FEP) can be used in principle to evaluate these
contributions by mutating residues involved in binding. In this work,
we use FEP to quantify the role of aromatics in the affinity of
Bacillus thuringiensis phosphatidylinositol-specific phospholipase C
(BtPI-PLC), phospholipase A2 (PLA2) from venom of the cobra (Naja naja
atra) and Neutrophil serine proteases Proteinase 3 (PR3) for
phosphatidylcholine-containing bilayers. We selected aromatic amino
acids located at various positions in the interfacial binding sites of
these peripheral proteins and mutated them to alanine to evaluate their
contributions in membrane binding. Earlier computational and
experimental work identified some of the tyrosines to mediate
cation-Ï€ interactions with choline groups from the
phospholipids, and others to partition at the interface without
mediating cation- π interactions in BtPI-PLC. The
cation-Ï€ interactions are less evident for
tryptophan and phenylalanine. The FEP results show significantly
different contributions for the aromatics involved in the
cation-Ï€ interactions than those without
cation-Ï€ interactions but partitioning in the head
group region. Tyrosine and phenylalanine contribute favorably around
1.0 Kcal/mol when partitioned at the phosphate plane and this
contribution increase to (2.0 - 2.5 )Kcal/mol and (2.5 - 3.0) Kcal/mol
respectively when mediating cation-Ï€ interactions.
The contribution from tryptophan is more favorable (around 2.0
Kcal/mol) due to its bigger size when partitioned at the phosphate
plane and increases with the depth of insertion to reach the same value
as for cation-Ï€ interactions where it contribute 3.0
– 3.5 Kcal/mol. Our results are in good
agreement with available experimental data and in particular with the
measured changes in the equilibrium dissociation constants of the
mutant compared to the wild type protein.
KM19
Is Your Mechanism Correct?
Kathrin H. Hopmann
Hylleraas Centre for Quantum Molecular Sciences, Dept. of
Chemistry,
UiT- The Arctic University of Norway, N-9037 Tromsø, Norway
Email: kathrin.hopmann@uit.no, Web: site.uit.no/CHOCO
Reaction pathways and properties of relatively large chemical systems
can nowadays be modelled with reasonable speed and good accuracies
[1,2]. However, mechanistic computations are often based on model
substrates, and the proposed pathways are not always validated against
known experimental information. Here I will show how the computation of
known selectivities of real substrates can help to establish the
validity of proposed mechanisms [3,4].
Left: Current computational methods are able to treat realistic systems
with good accuracies (from ref. 1a). Right: Computationally proposed
mechanisms require verification to ensure the validity.
References
- a) K. H. Hopmann, How accurate is DFT for iridium-mediated
chemistry, Organometallics 2016, 35, 3795, b) K.H. Hopmann, Quantum
chemical studies of asymmetric reactions: Historical aspects and recent
examples, Int. J. Quantum Chem. 2015, 115, 1232.
- K. H. Hopmann, Iron-Brønsted-acid-catalysed
asymmetric
hydrogenation: Mechanism and selectivity-determining interactions,
Chem. Eur. J. 2015, 21, 10020.
- G. R. Morello, H. Zhong, P. J. Chirik, K. H. Hopmann,
Cobalt-catalysed alkene hydrogenation: A metallacycle can explain the
hydroxyl activating effect and the diastereoselectivity, Chem. Sci.
2018, In Press.
- G. R. Morello, K. H. Hopmann, A dihydride mechanism can
explain
the intriguing substrate selectivity of iron-PNP-mediated
hydrogenation, ACS Catal. 2017, 7, 5847.
KM20
Computational insights into the decomposition pathways of
Ru-based olefin metathesis catalysts
Marco
Foscato, Wietse Smit ,Giovanni Occhipinti, Vidar R. Jensen
Department of Chemistry, University of Bergen, Norway
Olefin metathesis is “one of
organic chemistry’s most important
reactions†and the development of such fundamental
tool granted the Nobel Prize in Chemistry for 2005.[1]
In this reaction a transition metal complex catalyzes the formation of
carbon-carbon double bonds between two alkene substrates making the
synthesis of complex olefins “simple to
use (stable in air), efficient, and environmental
friendly.â€[1]
However, despite these promises, implementations of olefin
metathesis processes in the pharmaceutical industry highlight low
productivity as a major challenge.[2] Drug
synthons are, in fact, usually highly functionalized molecules often
characterized by polar and acid-base groups that trigger catalyst
decomposition.
To gain the insight necessary to understand and prevent
catalyst decomposition, we investigated the reaction network of one of
the most popular Ru-catalyst for olefin metathesis
(Hoveyda-Grubbs’s second generation
catalyst). Computational studies and experiments allowed the
exploration of the complicated reaction network that surrounds the
known olefin metathesis reaction mechanism. The identification of a
pivotal decomposition intermediate led to the discovery of reaction
pathways transforming the metathesis catalyst into the deleterious
olefin isomerization catalyst.[3] Moreover,
deprotonation of the metallacyclobutane species, which is the key
intermediate in olefin metathesis, could explain the reduction of
turnovers in presence of Brønsted base,[4]
and bimolecular coupling of two Ru-alkylidenes was also shown to
generate metathesis inactive species.[5]
This knowledge clarifies the weaknesses of the vulnerable
species involved in productive olefin metathesis reaction, and provides
the basis for catalyst redesign.
References
- The Nobel Prize in Chemistry 2005. NobelPrize.org. Nobel
Media AB 2018. https://www.nobelprize.org/prizes/chemistry/2005/summary
- C. S. Higman, J. A. M. Lummiss , D. E. Fogg, Angew.
Chem. Int. Ed. 2016, 55, 3552-3565.
- J. Engel, W. Smit, M. Foscato, G. Occhipinti, K. W.
Törnroos , V. R. Jensen, J. Am. Chem. Soc.
2017, 139, 16609-16619.
- G. A. Bailey, J. A. M. Lummiss, M. Foscato, G. Occhipinti,
R. McDonald, V. R. Jensen , D. E. Fogg, J. Am. Chem. Soc.
2017, 139, 16446-16449.
- G. A. Bailey, M. Foscato, C. S. Higman, C. S. Day, V. R.
Jensen , D. E. Fogg, J. Am. Chem. Soc. 2018,
140, 6931-6944.
KM21
Platform for Ontology Engineering and Evolution for Nano and Quantum Technologies
Axel Peter MUSTAD, Keeper Layne SHARKEY
Nordic Quantum Computing Group AS (NQCG), Oslo, Norway
Active direct communication with a uniquely recognizable language is
fundamental to knowledge transfer [1] between humanistic entities.
Building a standardized community with an expert level understanding of
rules for organizational classes of things, e.g., complex functional
nano-materials [2], quantum circuit protocols [3] from the emerging
field of quantum information theory, to the creation of various plasmas
[4], or even reinventing the shape of the periodic table of the
elements [5], is achieved by creating an effective ontology. The agreed
nomenclature is key to the growth of a healthy economic and scientific
sector as demonstrated by International Union for Pure and Applied
Chemistry (IUPAC) for the chemical industry starting in the early
1900’s [6] through the Gene Ontology Consortium in the 21st
century [7]. Integrating an ontology and managing it is a critical step
in increasing humans’ ability to solve advanced questions and
problems with a continually growing complexity; for instance, disease
discovery and verification in the medical field [8]. Through advanced
algorithms and artificial intelligence new ontologies have been
achieved by quantifying vast amounts of data and information. Nordic
Quantum Computing Group (NQCG) [9] has been directly involved with
coalescing nano-specific data sources [10].
Realizing a nano- and quantum-specific ontology (NaQuOnt) is central to
the mission of NQCG which aims to enable participation by forming
strategic partnerships with various academic and private establishments
globally to provide an open-source Software as a Service (SaaS).
NQCG’s core activity proposal is to engineer the implementation
of a suitable, web-based browsing tool that acts as a communication
forum and exploits machine learning with the use of Web Ontology
Language (OWL2), a standard language for data processing of the World
Wide Web Consortium (W3C) [11], in conjunction with Stanford’s
Protégé [12]. Validation of the NaQuOnt requires
interaction with leading researchers and innovators in the field;
current partners includes Google [13] and their development of quantum
computing capabilities allows for NQCG’s experimentation. To
emphasis, current quantum computing technologies are accelerating
computational quantum chemistry and will impact new material
discoveries and properties [14] which will directly impact the need for
said ontology. Integration of applications and software systems for the
resulting ontological capabilities is implemented across global
industry value chains in technology partnership with SAP’s
enterprise resources planning management systems [15]. Industry-quality
ontology standards created by NQCG and partners will further promote
global safety and compliance regulation, including risk analysis of the
developed technologies and consequently accelerating the maturation of
the technologies and the human experience of quantum chemistry.
References:
- Jasimuddin, S. M. and Zhang, Z. J. Operational Research Soc. 60, 706-716 (2009)
- Himanen, L., et. al., Computational Materials 4, 52 (2018)
- Pino, H., et. al., Quantum Science and Technology 3, 025001 (2018)
- Stockman, M., et. al. J. Opt 20, 043001 (2018)
- Grochala, W. Foundational Chemistry 20,191-207 (2018)
- https://iupac.org/
- http://www.geneontology.org/
- https://www.snomed.org
- http://nqcg.com/
- Karcher, S., et. al. NanoImpact 9, 85-101 (2017)
- https://www.w3.org/TR/owl2-overview/
- https://protege.stanford.edu/
- https://ai.google/research/teams/applied-science/quantum-ai/
- Reiher, M. et. al. PNAS 114, 7555-7560 (2017)
- https://www.sap.com/index.html
MK - Makromolekyl- og
kolloidkjemi
MK1
Nanoparticle delivery systems for antimicrobial peptides
Martin Malmsten
Department of Pharmacy, University of Copenhagen, DK-2100 Copenhagen, Denmark,
Department of Pharmacy, Uppsala University, P.O. Box 580, SE-752 32 Uppsala, Sweden
Presenting author email: martin.malmsten@sund.ku.dk
Due to rapidly increasing resistance development against conventional
antibiotics, finding novel approaches for the treatment of infections
has emerged as a major health issue. Antimicrobial peptides (AMPs) have
attracted interest in this context, and there is by now a considerable
literature on the identification such peptides, as well as on their
optimization to reach potent antimicrobial and anti-inflammatory
effects at simultaneously low toxicity against human cells. In
comparison, delivery systems for antimicrobial peptides have attracted
considerably less interest. However, such delivery systems are likely
to play a key role in the development of potent and safe AMP-based
therapeutics, e.g., through reducing chemical or biological degradation
of AMPs either in the formulation or after administration, by reducing
adverse side-effects, by controlling AMP release rate, by promoting
biofilm penetration, or through achieving co-localization with
intracellular pathogens.
Here, an overview is provided of some of our recent work on delivery
systems for antimicrobial peptides, including polymer nanogels[1,2],
mesoporous silica[3], nanoclays/nanosheets[4], and quantum dots, with
special focus on AMP-carrier interactions, as well as consequences of
these for membrane interactions, as well as for antimicrobial and
related biological effects of AMP-containing formulations.
Figure 1. Nanoparticulate drug delivery systems provide various advantages for antimicrobial peptides.
References
- R. Nordström, R., et al. J. Colloid Interface Sci. 2018, 513, 141.
- S. Singh, et al., ACS Appl. Mater. Interfaces 2017, 9, 40094.
- K. Braun, et al. J. Colloid Interface Sci. 2016, 475, 161.
- S. Malekkhaiat Häffner, et al. Phys. Chem. Chem. Phys. 2017, 19, 23832.
MK2
Microencapsulation and Controlled Release of Active Ingredients in Food, Feed and Personal Care Products.
Wilhelm R. Glomm, Peter Molesworth, Eugenia Sandru, Le Thuy Truong, Ruth Schmid, Heidi Johnsen
Polymer
Particles and Surface Chemistry Research Group, Department of
Biotechnology and Nanomedicine, SINTEF Industry, Trondheim, Norway
Correspondence: wilhelm.glomm@sintef.no
Microencapsulation is a process whereby an active ingredient –
gas, liquid or solid – is wrapped within a polymeric coating,
forming small particles, typically in the micrometer range. The polymer
acts as a barrier, isolating and protecting the active ingredient from
the external environment, and can dissolve or disintegrate through
specific stimulus, releasing the microcapsule content under specified
conditions. Depending on the active ingredient and the intended
application, microencapsulation is used to protect sensitive
ingredients such as vitamins from oxidation, increase
solubility/compatibility, mask unwanted taste and smell, increase
bioavailability, promote gradual or triggered release and more.
The Polymer Particles and Surface Chemistry Research Group at SINTEF
Industry is working with tailored encapsulation and release for a range
of applications and markets, using methods such as interfacial
polymerization, spray drying and complex coacervation. In this talk,
requirements for matching wall materials, microencapsulation method and
release profile will be discussed for several active ingredients used
in food, feed and personal care products.
MK3
Ultrafast DNA sequencing enabled by novel Ugelstad bead technology.
Synne Larsen(a),
Geir Fonnum(a), Talha Gökmen(a), Maarten Bruinsma(a), Lene
Husabø(a), Nini Hofsløkken Kjus(a), Grete Modahl(a),
Astrid Molteberg(a), Wolfgang Hinz(b), Prasanna Thwar(c)
a) Svelleveien 29, 2004 Lillestrøm, Norge
b) 246 Goose Lane, Guilford, CT, 06437, USA
c) 180 Oyster Point South San Francisco, CA 94080 USA
E-mail: Synne.larsen@thermofisher.com
DNA sequencing has become a disruptive tool in human diagnostics.
However, the high cost delayed and almost excluded the method to be
used in healthcare. Ion Torrent (part of Thermo Fisher Scientific) has
developed a next generation sequencing (NGS) method where their
semiconductor technology enables massive parallel sequencing, thereby
solving the cost challenge.
The actual sequencing occurs in a semiconductor chip, with millions of
micro-sized wells. The presence of one hydrogel bead in
each well are crucial for anchoring the genetic material within the
wells. Nucleotides are flooded over the chip and binds to the unpaired
nucleotides on the single stranded DNA on the bead. When a nucleotide
binds, a proton is released and gives a positive signal. Ion Torrent
were successful in the solution, but lacked a bead technology to secure
the required number of DNA in each well.
The beads must be highly monodisperse, with a tailor-made matrix that
is compatible with DNA and PCR. The talk will present how Thermo Fisher
Scientific Norway developed a novel seeded polymerization process for
custom-production of hydrogel beads for Ion Torrent, utilizing Ugelstad
technology. The traditional Ugelstad technology is limited to organic
soluble monomers, thus resulting in hydrophobic beads. To be able to
make not only hydrophilic, but hydrogel beads, new innovative solutions
were required.
Today Ion Torrent sequencers are the second most used sequencing
platform in the world and number one in oncology. The invention of the
tailor-made hydrogel beads was awarded both the Norwegian Research
Council Innovation Award and the Norwegian Tech Award in 2017.
MK4
Polymer films as matrix in drug delivery systems.
Ingunn Tho
School of Pharmacy, University of Oslo, Norway
Email: Ingunn.tho@farmasi.uio.no
Oral wafers or orodispersible films have become popular as drug
delivery systems, since they offer an alternative way to administer
drugs by melting or dispersing rapidly on the tongue without additional
water. Other types of film formulations are mucoadhesive systems that
will stick to the mucosa assuring prolonged contact time, which can be
utilized to improve absorption of the drug across the mucosa, either
the buccal mucosa, vaginal mucosa or intestinal mucosa.
Orodispersible films are usually prepared by the solvent-casting method
from a viscous polymer solution of a water-soluble polymer, such as
hydroxypropyl methylcellulose (HPMC), pullulan, starch or polyvinyl
pyrrolidone (PVP). The dry film is formed upon evaporation of the
solvent. In more advanced systems, the polymer film serve as a matrix
for nanoparticles, whose purpose might be to solubilize poorly soluble
drug, control the drug release, stabilize instable drugs or simply mask
the bitter taste of the drug. A few additional excipients are added to
the formulation in order to make it more palatable and patient
friendly. Typically, a plasticizer (e.g. glycerol, PEG), an aroma
and/or a sweetener and sometimes a filler to increase the content of
solid material in the dry film.
We work with various types of film formulations with and without
nanoparticles [e.g. 1-4], and some recent examples will be discussed in
the presentation.
References
- L.Roque, et al., Eur. Polym. J. 104 (2018) 19-31
- L.Roque, et al., Bioinspir. Biomim. 13 (2018) 055001
- J.Alopaeus et al., 2018 (submitted)
- J.Alopaeus et al. 2018 (in preparation)
MK5
Role of protein self-association on DNA condensation and nucleoid stabilization in a bacterial cell model
Rita S. Dias
Department of Physics, NTNU Norwegian University of Science and Technology, Trondheim, Norway
Bacterial cells do not have a nuclear membrane that encompasses and
isolates the genetic material. In addition, they do not possess histone
proteins, which are responsible for the first levels of genome
condensation in eukaryotes. Instead, DNA condensation in bacterial
cells is driven (at least partially) by DNA-binding proteins and
macromolecular crowding. Yet, some of these proteins also bind to RNA,
a component of ribosomes, which are present in large concentrations in
the cytosol of the cells.
In this work we use Monte Carlo simulations and a simple coarse-grained
bacterial cell model, enclosing a (bead and spring) model DNA, protein
dimers and crowding agents, to study the role of protein
self-association (a characteristic that some protein classes possess)
on DNA condensation and on the stability of the DNA-protein complex,
towards protein-competitive binding to the crowding agents. The results
of the simulations are in good agreement with exclusion dye experiments
conducted to probe synergism and competition effects of DNA
condensation by binding agents in the presence of crowding agents with
different characteristics.
MK6
On the Cooperativity during Melting and Molecular Exchange in Micelles with Crystalline Cores
Nico König, Lutz Willner, and Reidar Lund
Departmewnt of Chemistry, University of Oslo
Molecular exchange processes are important equilibration and transport
mechanisms in both synthetic and biological self-assembled systems such
as micelles, vesicles and membranes. Still these processes are not
entirely understood, in particular the effect of crystallinity and the
interplay between cooperative melting processes and chain exchange.
Here we focus on a set of well-defined polymer micelles formed by
binary mixtures of poly(ethylene oxide)-mono-n-alkyl-ethers (Cn-PEO)
which allows the melting point to be tuned over a wide range. We show
that while the melting transition is cooperative in the confined 2-3 nm
micellar core, the exchange process is widely decoupled and unimeric in
nature. As confirmed by calorimetry (DSC), the total
activation energy below the melting point is the sum of enthalpy of
fusion, and the corresponding activation energy in the melt state. This
suggests that while the crystallization process is cooperative,
a "single chain melting process" preludes the molecular diffusion
process during chain exchange.
MK7
Probing the Structure of Lipid Bilayers and their Interaction with
Indolicidin using Small Angle X-ray and Neutron Reflectivity methods
Josefine Eilsø Nielsen, Victoria Ariel Bjørnestad,
Abdullah Lone, Håvard Jenssen, Tania Kjellerup Lind,
Marité Cardenas and Reidar Lund
Department of Chemistry, University of Oslo
Antibiotic resistance is one of the biggest threats to global health,
according to WHO. AMPs seem to be able to evade much of the bacterial
resistance mechanisms and are therefore promising candidates for future
antibiotics. Instead of blocking specific biochemical pathways as most
available antibiotic agents today, most AMPs act physically on the
cytoplasmic membrane itself.[1-2] The precise microscopic mechanism for
the disturbance of the membrane has not fully been proven but several
theories has been suggested including membrane deformation and pore
formation. Here we have used state of the art neutron and x-ray
scattering techniques to investigate the microscopic mechanism of
action of AMPs with model bacterial membranes. SAXS measurements on a
model peptide, Indolicidin together with lipid vesicles has shown that
Indolicidin interacts with the membrane. Based on analysis of the
results we can see that the peptide does not seem to affect the
structure of the bilayer significantly but situates in the interface
between the lipid head group and the tail in the outer leaflet in the
bilayer.[3] This causes and slight alteration in the lipid chain
packing as seen by calorimetry but does not lead to any significant
membrane thinning or similar. This is further confirmed by Neutron
Reflectivity and Atomic Force Microscopy on planar supported
bilayers.[4] Combining these techniques has given us a new important
insight into how the peptide interact with the bilayer. Rather than
supporting any specific structural model, we speculate that the
mechanism is compatible with the disordered model proposed by Wimley.[5]
References
- Hancock, R. E.; Rozek, A., FEMS microbiology letters 2002, 206 (2), 143-149.
- Jenssen, H.; Hamill, P.; Hancock, R. E., Clinical microbiology reviews 2006, 19 (3), 491-511.
- Nielsen, J. E.; Bjørnestad, V. A.; Lund, R., 2018, (in preparation)
- Nielsen, J. E.; Bjørnestad, V. A.; Lone, A.; Lind, T. K.; Jenssen, H.; Cardenas, M; Lund, R., 2018, (in preparation)
- Wimley, W. C., ACS chemical biology 2010, 5 (10), 905-917."
MK8
Living apart together: tuning supramolecular assembly to create functional nanostructured soft materials.
Christian Sproncken, Hande Cingil, Antonio Aloi, Neus Vilanova,
Isja de Feijter, Gijs ter Huurne, Martijn Gillissen, Lafayette de
Windt, Anja Palmans, Bert Meijer, Ilja Voets
TU- Eindhoven, NL
Self-organization provides a fast, efficient, and low-cost pathway to
functional and responsive hierarchically structured materials that are
difficult if not impossible to prepare by other means. I will focus on
our recent work in this area, wherein we develop novel approaches to
achieve a high level of control over intra- and interparticle
interactions to modulate assembly pathways and precisely direct the
microstructure and properties of the final material [1-10] targeting
temporally programmable assembly [9] and novel applications [10].
First, I will discuss how the hydrogen-bond driven assembly in solution
of C3-symmetrical discotics based on benzene-1,3,5-tricarboxamides
(BTAs) can be utilized to prepare a wide range of nanostructured
materials [1-5], ranging from stiff, helical, one-dimensional fibers
[1] to photosensitive Pickering emulsions stabilized by supramolecular
colloids [6-7]. I will showcase how (macro)molecular structure and
physico-chemical factors impact the hierarchical organisation of the
materials [4-5], which is crucial to further their application
potential in catalysis, sensing, drug delivery, and coating technology.
Second, I will address electrostatically driven assembly of block
copolymers into so-called complex coacervate core micelles (C3Ms)
composed of a mixed core of polyelectrolyte blocks and a corona
consisting of neutral solvent-swollen blocks [8-10]. Interestingly,
C3Ms are multi-responsive nanostructures that readily adapt to changes
in charge stoichiometry, pH, ionic strength, as well as external cues,
like temperature and light, provided suitable (co)polymers are
selected. An overview will be presented of recent work on various types
of C3Ms developed for specific biomedical and materials science
applications demonstrating e.g. temporally programmed association and
cargo release [9], as well as inhibition of ice recrystallization [10].
References
- Gillissen, M. A. J.; Koenigs, M. M. E.; Spiering, A. J. H.;
Vekemans, J. A. J. M.; Palmans, A. R. A.; Voets, I. K.; Meijer, E. W.
JACS, 2014, 136, 336.
- M. A. J. Gillissen, T. Terashima, E. W. Meijer, A. R. A. Palmans and I. K. Voets Macromolecules, 2013, 46, 4120.
- P. J. M. Stals, M. A. J. Gillissen, T. F. E. Paffen, T. F. A. de
Greef, P. Lindner, E. W. Meijer, A. R. A. Palmans, and I. K. Voets
Macromolecules, 2014, 47, 2947.
- G. M. ter Huurne, M. A. J. Gillissen, A. R. A. Palmans, I. K. Voets and E. W. Meijer Macromolecules, 2015, 48, 3949.
- Ter Huurne, G. M.; de Windt, L. N. J.; Liu, Y.; Meijer, E. W.; Voets I. K.; Palmans, A. R. A. Macromolecules 2017, 50, 8562-8569
- I. de Feijter, L. Albertazzi, A. R. A. Palmans, I. K. Voets, Langmuir, 2015, 31, 57.
- N. Vilanova, I. de Feijter, A. J. P. Teunissen and I. K. Voets, Nature Scientific Reports, 2018, 8, 1271.
- A. Aloi, C. Guibert, L. L. C. Olijve, I. K. Voets, Polymer, 2016, 107, 450
- H. E. Cingil, N. C. H. Meertens and I. K. Voets Small, 2018, 1802089, 1.
- C. C. M. Sproncken, R. Surís-Valls, H. E. Cingil, C.
Detrembleur, I. K. Voets, Macromol. Rapid Commun., 2018, 39, DOI:
10.1002/marc.201700814
MK9
Lipid nanotubes: a possible route to protocell formation and growth
Elif Senem Koksal, Susanne Liese, Ilayda Kantarci, Ragni Olsson, Andreas Carlson, Irep Gözen
University of Oslo
Membrane-enclosed cellular compartments create spatially distinct
microenvironments which confine and protect biochemical reactions in
the cell. On the early Earth, the autonomous formation of compartments
is presumed to have enabled encapsulation of nucleotides, satisfying a
starting condition for the emergence of life. Recently, surfaces have
become into focus as potential platforms for the self-assembly of
prebiotic compartments, as notably enhanced vesicle formation was
reported in the presence of solid interfaces. The detailed mechanism of
such formation at the mesoscale however is still under discussion.
Here we report on the spontaneous transformation of lipid reservoirs on
solid substrates to unilamellar membrane compartments through a
sequence of topological changes, proceeding via a network of
interconnected lipid nanotubes. We show that this transformation is
entirely driven by surface-free energy minimization and does not
require hydrolysis of organic molecules, or external stimuli such as
electrical currents or mechanical agitation. The vesicles grow by
taking up the external fluid environment, and can subsequently separate
and migrate upon exposure to hydrodynamic flow. This may explain, for
the first time, the details of self-directed transition from weakly
organized bioamphiphile assemblies on solid surfaces to protocells with
secluded internal contents.
MK10
Applied colloid chemistry - research in an industrial setting
Rolf Andreas Lauten
Borregaard
Dispersants and/or viscosity modifiers is one of the major application
areas for some of the chemicals produced at the Borregaard Biorefinery
in Sarpsborg. For development of existing and new application areas, an
understanding of the requirements in both the end application, but also
physical characteristics of the dispersants themselves are important.
The latter can be a challenge for a poorly defined natural polymer.
Examples illustrating the use within cement chemistry and gold leaching
will be given and discussed in relation to dispersant characteristics.
MK11
Paint is not just paint – Protective coatings for the wind power industry.
Jacob Stensgaard Diget
Jotun A/S
Jotun, a paint supplier with over 10,000 employees world-wide, are
expanding their portfolio to protective coatings for wind-turbine rotor
blades. The work associated with the development of a protective
coating for the leading edge of a wind turbine rotor blade will be
presented. The leading edge is exposed to some of the harshest
environments with raindrop impingement at rotor blade tip speed of up
to 500 km/hr. To withstand this, a special binder system (polymer
network) is needed along with a “perfect” (smooth and void
free) surface.
MK12
Multifunctional Nanomaterials – at the junction of polymer science and colloid chemistry.
Sulalit Bandyopadhyay, Jibin Antony, Karthik Raghunathan, Anuvansh Sharma
NTNU
The interdisciplinary nature of nanoscience has shown that combination
results in unprecedented nanomaterial properties that not only
represent that of the counterparts but exceed them. This has made
synthesis and properties optimization of polymer based nanomaterials a
fascinating field that requires knowledge at the intersection of
polymer science and colloidal chemistry. Such nanomaterials are capable
of performing multiple functions, for instance; respond to stimuli,
encapsulate cargo molecule, release cargo molecule in a controlled
fashion - all from a single platform. However, when it comes to
combination of two different kinds of nanomaterials, the emergent
nanomaterial loses potential in its desired applicability.
Here, we will discuss how concepts borrowed from surface and colloid
chemistry may be used to control the physico-chemical properties of
metallic nanoparticles (NPs) synthesized via thermal decomposition
method (Fe NPs) and solution based seeded-growth method (Au NPs).
Thereafter, the effect of varying reaction parameters such as mole
ratios of monomers, surfactant concentration on the physico-chemical
properties of poly(N-isopropyl-acrylamide) (pNIPAm) based hydrogels
will be discussed. One manifestation of combination of such hydrogels
and metallic NPs will be highlighted in drug delivery applications.
Tuning the release of active cargo depends not only on material
properties but also on the effect of external stimuli such as
temperature, pH and so on. The potential of such smart nanomaterials
will be shown in other environmental applications such as in
hydrological tracing, water management and others. The end-use of such
multifunctional nanomaterials depends on modulation of their
physico-chemical properties that rests back on knowledge utilization of
synthesis and functionalization.
MA - Matkjemi
MA1
Fisk, oppdrettsfisk og helse.
Helle Katrine Knutsen
Avdeling for Miljøeksponering og –epidemiologi, Folkehelseinstituttet, Oslo
Abstract mangler.
MA2
Overvåkningsprogrammer for oppdrettsfisk.
Rita Hannisdal
Avdeling Sjømat i Modellsystem, Havforskningsinstituttet, Bergen
Abstract mangler.
MA3
Hvordan skal vi forholde oss til at kjemiske analysemetoder blir stadig mer følsomme?
Gjermund Vogt
Fagsjef Kjemi, Eurofins Norge, Moss
Abstract mangler.
MA4
Fate of the antioxidant ethoxyquin from feed to salmon filet:
application of liquid chromatography coupled to ion mobility and high
resolution mass spectrometry.
Sylvain Merel
Avdeling Trygt Fôr, Havforskningsinstituttet, Bergen
Ethoxyquin is commonly present in fish feed and acts as an antioxidant.
The quick oxidation of ethoxyquin prevents the oxidation of other
components responsible for the nutritional quality of the feed.
Identifying the transformation products of ethoxyquin and examining
their occurrence and transfer from fish feed to salmon filet is
important for risk assessment purposes. Previous research relying on
chemical bench-scale oxidation and the analysis of fish feed allowed
identifying 37 transformation products of ethoxyquin. Therefore, this
study aimed at extending the current knowledge to the identification of
transformation products in fish muscle.
To achieve this objective, salmons were fed during 90 days with
fortified feed containing ethoxyquin at 0.5 mg/kg, 119 mg/kg, and 1173
mg/kg. In addition, the occurrence of ethoxyquin and its transformation
products was also assessed in fillets from 12 commercial Norwegian
farmed salmon. All salmon filets were extracted in acetonitrile and
analyzed by liquid chromatography with travelingwave ion mobility
spectrometry coupled to high resolution mass spectrometry. Salmon
filets from the feeding trial showed the occurrence of ethoxyquin along
with 23 transformation products resulting from dimerization,
oxygenation,cleavage, cleavage combined with oxygenation, cleavage
combined with conjugation, and other alterations. Among them, 10 were
characterized for the first time. In filets of farmed salmon intended
for human consumption, ethoxyquin was detected in 75% of the samples.
In addition, 24 transformation products were also detected with a
frequency ranging from 8% to 100%. In all salmon filets from both the
feeding trial and fish farms, transformation products resulting from
dimerization were by far the most abundant. In particular, the
ethoxyquin dimer 1,8-EQDM was the main transformation product.
Finally, ion mobility spectrometry provided additional confidence for
compound identification during screening analysis. In complex matrices
or when the abundance is too low to allow the detection of
characteristic fragments, collision cross section with a 2% deviation
lowers potential interferences and false positives. The current study
allowed a comprehensive knowledge of the fate of ethoxyquin from fish
feed to salmon filet, and brought the total number of transformation
products identified to 47.
MA5
Miljøeffekter av kjemikalier brukt mot lakselus.
Renée Katrin Bechmann
IRIS, Stavanger
Lakselus er et problem for fiskehelsen. Selv om oppdrettsnæringen
bruker ulike metoder for å hindre at laks får lus, brukes
det fremdeles betydelige mengder kjemikalier som medisin. Laksen blir
behandlet enten med medisinfôr som inneholder pesticider, eller
det tilsettes kjemikalier i vannet for å bli kvitt lus. Rester av
medisinfôr eller kjemikalier fra badebehandling i merden eller i
brønnbåt slippes rett ut i havet. Kanskje blir
miljøet bare utsatt for kjemikaliene en kort stund, men i
områder med oppdrett kan krepsdyr og andre organismer utsettes
for et eller flere kjemikalier flere ganger. Målet i PestPuls
prosjektet er å skaffe ny kunnskap om hvordan den viktige
dypvannsreka Pandalus borealis kan påvirkes av kjemikalier brukt
i badebehandling av laks. Det er viktig å finne ut hvor
følsom dypvannsreka er for kjemikalier som slippes ut fra
oppdrettsnæringen, siden den er en nøkkelart i
økosystemet og spises både av fisk og folk. Vi har sjekket
hvordan korte, gjentatte eksponeringer for AlphaMax (deltametrin) og
Salmosan (azametifos) alene og i kombinasjon påvirker rekene.
AlphaMax behandlingsløsning for laks inneholder 2 mikrogram
deltametrin per liter. Tusen ganger fortynnet (2 ng/L deltametrin)
løsning drepte voksne reker på mindre enn ett døgn.
Da rekene ble eksponert kun to timer per dag i en uke overlevde de, men
de spiste betydelig mindre enn kontrollen og det var skade på
vevet i fordøyelseskjertelen. Eksponering for titusen ganger
fortynnet løsning i ett døgn ga endringer i adferd, men
ingen dødelighet. Rekelarvene var enda mer følsomme for
AlphaMax enn de voksne rekene. Det var høy dødelighet
etter to timer med 2 ng/L, og få av de som overlevde klarte
å svømme.
Salmosan behandlingsløsning for laks inneholder 100 mikrogram
azametifos per liter. Det var ingen dødelighet av voksne reker
som ble eksponert i ett døgn for tusen ganger fortynnet
løsning (100 ng/L), og heller ingen dødelighet etter to
timers eksponering per dag i en uke, men det var skade på vevet i
fordøyelseskjertelen. Det var ingen tydelig effekt på
overlevelse for rekelarver utsatt for 100 ng/L i to timer. Reker utsatt
for både Salmosan og AlphaMax hadde lignende responser som de som
kun fikk AlphaMax, men det var mer skade på
fordøyelseskjertelen til voksne reker i den kombinerte
eksponeringen.
Paramove behandlingsløsning for laks inneholder 1500 mg/L
hydrogen peroksid. Økt dødelighet og redusert spiserate
ble observert for voksne reker etter eksponering for tre to-timers
pulser med 1.5 mg/L. Økt dødelighet ble også
observert etter kun to timers eksponering for 15 mg/L.
Dødeligheten skjedde omtrent tre dager etter eksponeringen
(forsinket effekt). Tydelige skader på gjellene og lipid
peroksidering i fordøyelseskjertelen ble funnet etter en times
eksponering for 1.5 mg/L og 15 mg/L hydrogen peroksid.
Hovedkonklusjonen er at få timers eksponering for AlphaMax eller
Paramove førte til økt dødelighet av reker selv
ved tusen ganger lavere konsentrasjon enn det som brukes til å
behandle laksen.
PestPuls ledes av NORCE med NIVA (kjemi og modellering), Universitetet
i Leicester (genekspresjon), DEBtox Research (modellering) og Burridge
Consulting Inc. som partnere, i tillegg til en nasjonal
rådgivningsgruppe med representanter fra forvaltning, fiskeri og
oppdrett. Dette foredraget vil fokusere på effekter på
rekene i forsøk utført av NORCE i 2017 og 2018.
OR - Organisk kjemi
OR1
Harnessing Nature's Toolbox for Selective Halogenations: New
Concepts for an Old Problem.
Tanja Gulder
Biomimetic Catalysis, Department of Chemistry and Catalysis
Research Center, Technical University Munich, Lichtenbergstrasse 4,
85748 Garching, Germany.
Email: Tanja.Gulder@tum.de
Although the halogenation of organic molecules
is one
of the most widespread techniques for the functionalization of
substrates,
efficient catalytic methods for the selective (regio-, chemo-, and in
particular stereoselective) construction of carbon-halogen bonds are
rare. In
contrast, Nature has evolved different strategies to create
carbon-halogen
bonds in a highly effective and specific manner.
Our research therefore focuses on the
exploration of the mechanism as well as the structure-function
relationship of
such halogenation catalyzing enzymes. Based on these findings mild,
generally
applicable, and selective catalytic methods for the formation of
carbon-halogen
bonds (brominations, chlorinations and even fluorinations) are
developed
combined with their application to access medically relevant target
structures
in efficient ways. Selected examples of our group imitating
Nature’s concepts
of halogenation reactions will be presented, reaching from
biotransformation [1]
to λ3-iodane [2] and amine [3] mediated
transformations.
References
- a) Frank, A.; Seel, C. J.; Groll, M.; Gulder, T. ChemBioChem 2016,
17, 2028; b) C. J. Seel; A.
Králík, M.
H., A. Frank, B. Koenig, T. Gulder ChemCatChem
2018, online
available, doi.org/10.1002/cctc.201800886
- a) Fabry, D. C.;
Stodulski, M.; Hoerner, S.; Gulder, T. Chem.
Eur. J. 2012, 18, 10834; b) Stodulski, M.; Goetzinger,
A.; Kohlhepp, S. V.; Gulder, T. Chem.
Commun. 2014, 50, 3435; c) Ulmer, A.; Stodulski, M.;
Kohlhepp, S. V.; Patzelt, C.; Poethig, A.; Bettray, W.; Gulder, T. Chem. Eur. J. 2015,
21, 1444; d) Ulmer,
A.; Brunner, C.; Arnold, A. M.; Poethig, A.; Gulder, T. Chem.
Eur. J. 2016, 22, 3660; e) Arnold, A. M.; Ulmer, A.;
Gulder, T., Chem. Eur. J. 2016, 22,
8728; f) Kohlhepp, S. V.; Gulder, T. Chem.
Soc. Rev. 2016, 45, 6270; g) Patzelt, C.; Poethig, A.;
Gulder, T. Org. Lett. 2016, 18,
3466; h) Brunner, C.; Andries-Ulmer, A.; Kiefl, G. M.; Gulder,
T. Eur. J. Org. Chem. 2018, 2615; i) Andries-Ulmer, A.;
Brunner, C.; Rehbein, J.; Gulder, T. J.
Am. Chem. Soc. 2018,
accepted.
- A. M. Arnold, A. P., M. Drees, T. Gulder J.
Am. Chem. Soc. 2018, 140, 4344.
OR2
Catalytic Approaches for Simplifying Complex Molecule
Synthesis
Darren J. Dixon
Department of Chemistry, University of Oxford, Oxford, OX1
3TA, UK
Email: (darren.dixon@chem.ox.ac.uk)
Catalysts that provide new reactivity and stereocontrol in efficient
bond-forming reactions, are essential tools for converting low cost
starting materials into high value, structurally complex,
stereochemically defined product materials. In this presentation, new
families of metal-free and metal-rich cooperative catalysts and their
use in highly enantioselective C-C bond forming reactions and other
relevant transformations, will be described.
Their strategic application to the discovery of new one-pot reaction
cascade processes to generate novel, stereochemically defined scaffolds
and architectures useful for library and target synthesis will also be
discussed. Further application of selected methodologies as pivotal
carbon-carbon bond forming steps in the total synthesis of a range of
manzamine, aspidosperma, iboga, strychnos and daphniphyllum alkaloids
will then be discussed. These syntheses serve to illustrate how complex
molecular targets can be rapidly accessed when combinations of
catalyst-controlled reactions, one-pot multistep procedures and
powerful route-shortening cascades are designed into the overall
synthetic sequence.[1-10]
References
- F. Sladojevich, A. Trabocchi,
A. Guarna, D. J. Dixon, J. Am.
Chem. Soc. 2011, 133,
1710.
- M. Yu, C. Wang, A. F. Kyle,
P. Jakubec, D. J. Dixon, R. R. Schrock, A. H. Hoveyda, Nature,
2011, 479, 88.
- P. Jakubec, A. Hawkins, W.
Felzmann, D. J. Dixon, J. Am. Chem.
Soc. 2012, 134, 17482.
- M. G. Núñez, A. J. M. Farley, D. J.
Dixon, J. Am. Chem. Soc. 2013 135,
16348.
- I.
Ortín, D. J.
Dixon, Angew. Chem. Int. Ed. 2014, 53,
3462.
- A. D. Gammack Yamagata, S.
Datta, K. E. Jackson, L. Stegbauer, R.
S. Paton, D. J. Dixon, Angew. Chem. Int.
Ed. 2015, 54,
4899.
- R. De La
Campa, I.
Ortín, D. J.
Dixon, Angew. Chem. Int. Ed. 2015, 54,
4895.
- A. J. M. Farley, C.
Sandford, D. J. Dixon, J. Am. Chem. Soc.
2015, 137,
15992.
- J. Yang, A. J. M. Farley, D. J.
Dixon, Chemical Science, 2017, 8, 606.
- P. W. Tan,
J. Seayad,
D. J. Dixon, Angew.
Chem.
Int. Ed. 2016, 55,
13436.
OR3
Concise total synthesis of
(±)-dehaloperophoramidine
Peter Somfai
Center for Analysis and Synthesis, Department of Chemistry,
Lund University, Box 124, 22100 Lund, Sweden
Email: peter.somfai@chem.lu.se
Perophoramidine and
communesin F are
structurally related indole alkaloids. Dehaloperophoramidine is
synthetic analogue of perophoramidine lacking aromatic halogens. Our
study towards the total synthesis of dehaloperophoramidine have led to
the discovery of two novel domino processes that significantly improve
the efficiency of the total synthesis. The first domino process
encompasses four steps and resulted in the formation of ortho-amide.
The second domino was discovered through a careful examination of the
reactivity of ortho-amide, ultimately resulting in the target compound.
The vicinal quaternary stereocenters was installed early in the
synthesis by employing Overman’s
samarium mediated
reductive dialkyltion procedure. Herein is the total synthesis of
dehaloperophoramidine in eight step and 23 % overall yield. In
addition, our studies towards Strictamine and Cephalotaxine will also
be discussed.
References
- Popov, K.; Hoang, A.; Somfai, P. Angew. Chem. Int. Ed. 2016,
53, 1801.
UM - Uorganisk kjemi og
materialkjemi
UM1
Defect chemistry of functional oxides and their interfaces
T.
Bjørheim
University of Oslo
Defects affect and give rise to a wide range of functional properties in oxide ceramics. Li+, H+, O2- and H-
species enable macroscopic ionic transport while a variety of
aliovalent ionic defects act as strong charge carrier traps.
Higher-dimensional defects such as surfaces, grain boundaries, and
interfaces, being both structurally and chemically different to their
bulk counterparts, may similarly act as sinks for variety of charged
defects, in turn leading to charge-imbalance and deviations from local
electroneutrality in the form of space-charge regions. This
contribution explores the fundamental driving forces of defect
formation in functional oxides, and accumulation at interfaces, by
introducing the concepts of lattice site basicity and ion affinities
and show how these properties can be determined from first principles
calculations. The concept is applied to hydration of proton conducting
oxides, showing how the oxides’ chemistry affect the defect
chemistry of both bulk materials, their interfaces, and of
nanocomposites.
UM2
Bimetallic CoRe in APD silica aerogels for ammonia
decomposition.
Karsten Granlund Kirste*, Karina
Mathisen*, Laura
Torrente Murciano†,
Dragos Stoian¶,
Said Laassiri‡,
Justin
Hargreaves‡.
*Norwegian University of Science and Technology (NTNU),
N-7491 Trondheim, Norway.
†University
of Cambridge, UK.
¶Swiss-Norwegian
Beamlines, European
Synchrotron Radiation Facility, Grenoble, France.
‡University
of Glasgow, UK.
Silica aerogels are
ultra-porous amorphous materials consisting of interconnected silica
particles and mostly air. The silica matrix is porous and consisting of
micro- and mesopores, have a high surface area, low thermal
conductivity and exhibits interesting surface characteristics such as
hydrophobicity [1, 2]. Deposition and formation of nanostructures in
this 3D support may result in high dispersions. Growth limitations
caused by the support can greatly affect both reducibility and
reversibility of the metal phase, which will influence the reactivity
and economy.[3, 4].
Production of hydrogen
from ammonia is widely studied due to its high hydrogen content and
relative ease of storage. Ammonia decomposition is favoured by metal
supported on more or less porous carriers and for this purpose,
bimetallic phases of cobalt and rhenium supported on silica aerogel
have been synthesised through co-precipitation and then dried with the
ambient pressure drying method (APD). A range of CoRe@aerogels were
prepared and the activity towards ammonia decomposition depend greatly
on total metal loading, Co/Re-ratio and annealing. In situ XAS reveal
that reduction of both cobalt and rhenium occurs simultaneously during
H2-treatment and over a lower temperature range (300-350°C)
than their monometallic analogues, suggesting a synergistic effect
occurs promoted by the 3D support.
The CoRe@aerogel have a
high degree of Co-Re mixing with the major phase from EXAFS analysis
during pre-treatment and ammonia decomposition being small Co-Re
clusters (<1nm). Interestingly an increase in the he
multiplicity of a Co-O/N shell is observed during ammonia decomposition
indicative of surface adsorbed nitrogen or increased interaction with
the support.
References:
- S.
D. Bhagat, Y.-H. Kim, K.-H. Suh, Y.-S. Ahn, J.-G. Yeo and J.-H. Han, Microporous and Mesoporous Materials,
2008, 112, 504-509.
- J.
Fricke and T. Tillotson, Thin Solid Films,
1997, 297, 212-223.
- J.
de Graaf, A. J. van Dillen, K. P. de Jong and D. C. Koningsberger, Journal of Catalysis, 2001, 203, 307-321.
- D. B. Akolekar and
S. K. Bhargava, Journal of Molecular
Catalysis A: Chemical,
2005, 236, 77-86.
UM3
Performance of all-oxide thermoelectric generator enhanced by
high-temperature interfacial chemistry.
N.
Kanas, M. Bittner, T.D. Desissa, S.P. Singh, T. Norby, A.
Feldhoff, T. Grande, K. Wiik, M.-A. Einarsrud
NTNU
Thermoelectric generators (TEGs) represent an important and promising
technology enabling the direct conversion of heat to electric energy.
The traditional TEGs are composed of metals and applications are
limited to low and medium temperatures due oxidation and melting. For
high-temperature applications in ambient atmosphere all-oxide TEGs
represent an attractive approach, both due to their high stability and
low environmental impact.
To utilize the advantages of oxides at high temperature, thermoelectric
modules with direct p-n junction (Fig. 1a) was fabricated using p-type
Ca3Co4O9
(CCO) and n-type Ca0.931MnO3
(CMO). The
processing was conducted by spark plasma co-sintering, using tape
casted LaAlO3 (insulator) in between the two
conductors to partially
separate them. A layer of a p-type Ca3CoMnO6
(CCMO) was formed at the
interface between the p- and n-type materials during sintering. The
CCMO-phase showed unusually high Seebeck coefficient, beneficial for
the performance of the device.
In this contribution we will present our ceramic processing approach
and discuss the high- temperature chemistry of the material system. The
results from a successfully tested co-sintered TEG (Fig. 1 a and b)
will be presented in more detail, and possible reasons for the
unusually high power output will be discussed.
Fig. 1 a) Cross-section of the module with illustrated current and heat
flow; b) Electrical power output of the module
UM4
Surface modification of Ta3N5 nanotubes as photocatalyst for
photoelectrochemical water splitting.
Kaiqi
Xu1, Athanasios Chatzitakis1, Ingvild Julie Thue Jensen2,
Mathieu Grandcolas2, Truls Norby1*
(1) Centre for Materials Science and Nanotechnology,
Department of
Chemistry, University of Oslo, FERMiO, Gaustadalléen 21,
NO-0349
Oslo, Norway.
(2) SINTEF Industry, P.O. Box 124 Blindern, NO-0314 Oslo, Norway
*corresponding author truls.norby@kjemi.uio.no
Ta3N5
nanotubes (NTs) were
obtained from nitridation of Ta2O5
NTs, which were grown
directly on Ta foil through a 2-step anodization procedure. A
“waggling”
appearance close to the “mouth” of Ta2O5
NTs was
observed, and after nitridation, a unique mesoporous structure appeared
on the
walls of the Ta3N5 NTs. With
Co(OH)x
decoration, a photocurrent density as high as 2.3 mA/cm2
(1.23 V vs.
NHE) was reached under AM1.5G simulated solar light, however, the
electrode
suffered from photocorrosion. More stable photoelectrochemical (PEC)
performance was achieved by first loading Co(OH)x,
followed by
loading cobalt phosphate (Co-Pi) as double co-catalysts. The Co(OH)x/Co-Pi
double co-catalysts may act as a hole storage layer that slows down the
photocorrosion caused by the accumulated holes on the surface of the
electrode.
Optimized parameters, e.g. tuned deposition time, can further enhance
the PEC
performance and stability. The accumulated hydrogen production will be
measured
and quantified.
Figure 1. a) SEM
image of the “mouth” of Ta3N5 NTs, b) j-U curves of
Ta3N5
NTs with different surface modification and c) chronoamperograms
showing the
performance stability of different Ta3N5 NTs surface
modifications
Acknowledgement:
Financial
support from the Research Council of Norway (CO2BiOPEC project 250261)
is
acknowledged.
UM5
Can hydroxyl radicals travel far? Gas phase
transport and
detection after photocatalytic generation at TiO2 nanorods.
X. Sun,
K. Xu, A. Chatzitakis, T. Norby*
Department of Chemistry, University of Oslo, SMN, FERMiO,
Gaustadalléen 21, NO-0349 Oslo, Norway
Tel.: +47-22840654
truls.norby@kjemi.uio.no
The
quality of indoor environment is of great importance as it can
significantly
improve the human health, comfort and productivity [1]. Reactive
oxidising
species (ROS) can keep our air clean because they can non-selectively
oxidise a
wide range of air contaminants. Hydroxyl radicals are among the
strongest ROS
in the atmosphere and devices that can efficiently produce them can
become an
important class of air-cleaning solutions. TiO2
in the presence of
water and UV light has a unique ability to produce hydroxyl radicals
[2]. Here,
TiO2 nanorods (TNRs) are synthesised and used as
the photocatalyst
for hydroxyl radicals generation in a gas phase photoreactor, taking
advantage
of the water that is present in air. Therefore, air with controlled
relative humidity
(RH) levels is used as the water supply. The main aim is to study the
effectiveness of the radicals as a function of RH, as well as the
distance from
a model pollutant. It is also of interest to quantify the amount of
radicals
that are photocatalytically produced, bearing in mind that such a
system can
become a commercial cleaning device. To achieve the above, we studied
the
remote decolourisation of a solid Methylene Blue (MB) film and the
change in
conductivity of an electrochemical sensor, based on polyaniline (PANI).
We
observed a remote decolourisation efficiency of 26% under 80% relative
humidity
when the photocatalyst was placed 0.5 cm away from the MB film. This
efficiency
was reduced to 19% when the distance increased to 3 cm. Moreover, a
rate of 1012 radical
molecules per second were photocatalytically produced, which were
calculated by
the change in conductivity of the PANI sensor. Therefore, we hold it
likely
that hydroxyl radicals can travel far and generators with certain
requirements can be developed.
Figure 1: Effect of UV
light,
photocatalyst (TNRs) and its distance from the MB at 80% RH. Airflow
rate: 0.75 mL/min (a), digital image of a MB film used as the
colorimetric indicator (b), digital image of an interdigitated
electrode coated with PANI and used as an electrochemical sensor(c).
References
- S.W. Verbruggen, J. Photochem Photobiol. C, Photochem Rev,
24 (2015) 64.
- R. Andreozzi, et al., Catal Today, 53 (1999) 51.
Acknowledgement:
This work was
performed within MoZEES, a Norwegian Centre for Environment-friendly
Energy Research (FME), co-sponsored by the Research Council of Norway
(project number 257653) and 40 partners from research, industry and
public sector. We also acknowledge support from the Research Council of
Norway under the CO2BioPEC project 250261.
UM6
From inorganic chemistry to components for
Li-/Na-ion
batteries - insight from operando studies.
Helmer Fjellvåg
Department of Chemistry / SMN, University of Oslo
Inorganic materials chemistry plays a major role in development of
materials for Li- and Na-ion battery technology. Different requirements
apply depending on whether the material will enter as the electroactive
material in a cathode or an anode, or whether it takes a role as solid
state electrolyte. Improved materials are highly requested in order to
develop new technology that may provide higher energy capacities in
batteries for e.g. the transportation sector. In this talk some
principles related to chemical bonding, redox properties and atomic
arrangement will be discussed. The effect of nanostructuring of
materials will be demonstrated by examples. Information on how an
electrode material performs during cycling (charge/discharge) is
obtained from electrochemical data, however, we will show the
importance of performing operando studies by X-ray methods (home lab
and at synchrotrons) for gaining essential insight into mechanisms
related to redox, conversion and intercalation reactions, and thereby
reversibility and capacity fading.
UM7
High capacity magnesium batteries using
solvent-controlled
charge storage.
Lu
Wang a, Zhaohui Wang a,b,
Per Erik Vullum b,c,
Sverre Magnus Selbach a, Ann Mari Svensson a,
Fride
Vullum-Bruer a
a Department of Materials Science and
Engineering, Norwegian University of Science and Technology, NO-7491
Trondheim,
NORWAY
b
SINTEF
Industry, NO-7491
Trondheim, Norway
c Department of
Physics, Norwegian University of
Science and Technology, 7491 Trondheim, Norway
E-mail: fride.vullum-bruer@ntnu.no
The increased demand for
portable as
well as stationary energy storage has in the last few decades lead to
heavy
research efforts into a variety of battery chemistries. Li-ion
batteries is
still dominating the marked for small-scale electronics as well as
electric
vehicles. However, as more and more devices are electrified and the
demand for
tailored batteries increase, new chemistries become significantly more
important. One of the more promising new technologies is the Mg-ion
battery
(MIB) due to higher safety, chemical stability and a high natural
abundance in
the Earth’s
crust
(13.9% as compared to 7*10-4 % for Li). In
addition, Mg has a
theoretical volumetric capacity of 3833 mAh/cm3,
nearly twice that
of Li (2061 mAh/cm3), indicating the potential
of MIB to reach a
high volumetric energy density.1, 2 There are
however, several
issues hampering a fast development of MIBs, including finding suitable
electrolytes as well as good cathode materials capable of providing
high
specific capacity and power density, good rate performance, and
long-term
stability. And one of the main issues for the cathode materials is poor
diffusion of the divalent Mg ion in the host material.
Here, we present a Mg
battery using Mn3O4 as
the electrode material and Mg
metal as the counter electrode in a Mg organohaloaluminate electrolyte.
The
material has been investigated both as cathode and anode, simply by
changing
the cut-off voltage during electrochemical testing. The reversible
capacity
when Mn3O4 was used as
cathode reached ~580
mAh g−1
at a current density
of 15.4 mA g−1,
whereas a reversible
capacity of ~1800 mAh g−1
was obtained in an
anode configuration. As a cathode, the Mn3O4
showed
excellent cycling stability with no loss of capacity after 500 cycles
at a
current density of 770 mA g−1.
As an anode, the
stability was not as good and Mn3O4
retained 86% of its
initial capacity after 200 cycles. By careful investigation using
quantitative
kinetics analysis in addition to TEM with EDX and EELS it was found
that these exceptional
charge storage properties and high cycling stability are attributed to
highly
reversible interfacial reactions involving the electrolyte solvents.
Density
functional theory calculations were also conducted, which support these
findings.
Acknowledgements:
Norwegian Research Council for funding
(Grant
Number 221785). Computational
resources were provided by the Norwegian Metacenter for Computational
Science (NOTUR) through the project NN9264K and NTNU243.
References:
- M. Matsui, J. Power Sources, 196
(2011), 7048.
- C.
Ling, D. Banerjee and M. Matsui, Electrochim. Acta, 76 (2012), 270.
UM8
Electrospinning
of Ba0.85Ca0.15Zr0.10Ti0.90O3
nanofibers for
flexible nanogenerators.
Per
Martin Rørvik*, Mathieu Grandcolas, Christelle
Denonville, and Tor Olav Sunde
SINTEF, Forskningsveien 1, Oslo, Norway
*Corresponding Author: per.martin.rorvik@sintef.no
Certain piezoelectric applications require large-area and flexible
devices, for instance for energy harvesting from wearable devices [1]
or anti-fouling membranes for water treatment [2]. Electrospinning of
nanofibers into a mat is a useful method for fabrication of the active
piezoelectric material for such flexible devices. Here, nanofiber mats
of the lead-free piezoelectric oxide Ba0.85Ca0.15Zr0.10Ti0.90O3 (BCZT)
were made by electrospinning using an Elmarco NS needleless
electrospinner, followed by annealing in air. The BCZT mat was put
together with electrode polymer films into a flexible flat device and
mechanical energy harvesting was demonstrated by recording voltage
output when bending the device.
References
- Q. Yang, et al., Journal of Alloys and Compounds, 688,
1066-1071 (2016)
- J. Bae, et al., Chemical Engineering Journal, 307,
670–678 (2017)
UM9
Verification
of hierarchical porosity in CuSAPO-34 by in situ XAS, N2
adsorption
measurements and NOx removal.
Guro
Sørli*,Dragos Stoian†, Magnus
Rønning§,Karina Mathisen*
*Department of Chemistry, §Department of Chemical
Engineering,
Norwegian University of Science and Technology, 7491 Trondheim, Norway
†Swiss Norwegian Beam Lines, European Synchrotron Radiation
Facility, Grenoble, France
guro.sorli@ntnu.no
Removal of NOx from combustion processes made headlines in 2015
following the so-called “diesel-gate”, showing that
new
development and research concerning deNOx technology is still highly
topical1. Copper containing, microporous SAPO-34 has shown great
activity concerning selective reduction of NOx (NH3-SCR and HC-SCR) and
may be thought of as a new possible catalyst for NOx removal from
internal combustion engines2-6. These catalysts are known to suffer
from instability concerning copper addition and deactivation due to
coking. The goal of this project is to solve these challenges by
introducing mesopores to create so-called hierarchical CuSAPO-34 to
relieve the mass transfer issues.
Different structure directing agents have been used to obtain
hierarchical CuSAPO-34 and comparisons have been made with the
conventional microporous analogue. In-situ XAS data has been recorded
at the Swiss-Norwegian Beam Lines (SNBL, BM 31) at the ESRF in
Grenoble, France in order to obtain information about the reducibility
and size of copper clusters in the samples. Multivariate curve
resolution (MCR) analysis has been utilised to obtain reduction
profiles of copper. Results from in situ XAS analysis have been
correlated with BET surface area and BJH pore size distribution
measurements. In the presence of copper, the structure directing agent
(SDA)-pair diethylamine (DEA) + tetraethylenepentamine (TEPA) yields a
high degree of mesoporosity in CuSAPO-34, whereas the SDA-constellation
morpholine (MOR) + TEPA + cetyltrimethylammonium hydroxide (CTAOH)
yields mostly micropores. The pore distribution show a large number of
mesopores ranging from 30 – 200 Å in the former,
correlating with 100 m2/g external/meso area from the t-plot, not
present in the sample made with CTAOH. The introduction of mesopores
greatly affects the reducibility of copper during temperature
programmed reduction (TPR) by H2 (75%), as copper is completely reduced
at 490°C in the sample containing mesopores, but 20% CuI-O
remains
in the microporous sample, even at 700°C. The introduction of
mesopores is again reflected in the obtained copper particle sizes from
EXAFS analysis, as the mesoporous CuSAPO-34 hosts clusters of 14
Å (NCu-Cu = 8) whereas they are found to be 9 Å
(NCu-Cu =
6) in the sample with mainly micropores (the method of corrected
multiplicities were employed for the latter sample).
Employing HC-SCR deNOx as a model reaction, the introduction of
mesopores greatly improves the NOx conversion over the whole
temperature range (275- 500°C), but especially in the low
temperature range (<375°C). Whereas the hierarchical
CuSAPO-34
made with DEA-TEPA becomes active at 325°C reaching maximum
conversion of 67% at 400°C, the microporous
becomes active at 375°C and reaches maximum conversion of 52%
at
450°C. Clearly, altering the porosity of CuSAPO-34 has great
impact
on chemical and catalytic behaviour of the zeotype.
References
- R. Hotten, Journal, 2015.
- U. Deka, I. Lezcano-Gonzalez, S. J. Warrender, A. Lorena
Picone,
P. A. Wright, B. M. Weckhuysen and A. M. Beale, Microporous and
Mesoporous Materials, 2013, 166, 144-152.
- T. Jakobsen, Master Thesis, NTNU, 2014.
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C.
Lamberti, P. Beato, H. Falsig and S. Bordiga, Journal of the American
Chemical Society, 2016, 138, 12025-12028.
- M. Moliner, C. Martínez and A. Corma, Chemistry
of Materials, 2014, 26, 246-258.
- D. Wang, L. Zhang, K. Kamasamudram and W. S. Epling, ACS
Catalysis, 2013, 3, 871-881.
UM10
Coupled
hydration, cation reorganization and oxidation in MIEC perovskites.
Ragnar
Strandbakke*1, Tor S.
Bjørheim1, Aleksandra
Mielewczyk-Gryń2, Sebastian Wachowski2, Iga
Lewandowska2, Maria Gazda2,
María Balaguer3, Jose M. Serra3,
Magnus H. Sørby4,
Truls Norby1
- Department
of Chemistry, University of Oslo, FERMiO, Gaustadalléen 21,
NO-0349 Oslo,
Norway.
- Department
of Solid State Physics, Faculty of Applied Physics and Mathematics,
Gdańsk
University of Technology, Narutowicza street s11/12, 80-233 Gdańsk,
Poland
- Instituto
de Tecnología Química (Universitat
Politècnica de València - Consejo Superior
de Investigaciones Científicas), Avenida de los Naranjos
s/n.46022 Valencia,
Spain
- Department for Neutron Materials
Characterization, Institute for Energy Technology, NO-2027 Kjeller,
Norway
Tel.:
+47-22840660
ragnarst@smn.uio.no
Hydration of some La-containing Co-based
perovskites and double perovskites
is characterized by an initial fast mass gain followed by a much slower
and
larger weight increase. The latter process has equilibrium times of
typically
five days for a pre-calcined powder at 400°C, accounting for
around 80% of the
total weight gain. Dehydration exhibits the same initial fast weight
drop and a
following slow weight loss. We hypothesize that the initial hydration
changes
the lattice so as to induce a slower secondary process. If the
secondary
process was a second hydration, dehydration would still be fast when
water
vaour was removed. This is however not the case, hence the secondary
process
must reflect another reaction. The exchange of oxygen is pronounced in
these
mixed conducting cobaltites, and we believe that hydration changes the
degree
of cation order, which in turn alters oxidation thermodynamics.
In double
perovskites with the general formula AIAIIB2O6-δ,
oxygen vacancies are formed to compensate for the size mismatch between
Ba at AI
and a lanthanide (Ln) at AII. The result is an
increased Co radius,
which accounts for most of the size compensation. At 300°C,
tetragonal BaLaCo2O6-δ
(BLC) shows a δ of 0.1,[1] Ba1-xGd0.8-yLa0.2+x+yCo2
O6-δ
with x=y=0 (BGLC1082) shows a δ of 0.35 [2] and BaGdCo2O6-δ
(BGC) shows a δ of 0.5.[3] BGLC1082, exhibiting an intermediate oxidation of
Co with respect to BLC
and BGC, is shown to hydrate at high temperatures.[4] While hydration of BGC is limited, hydration of
BGLC decreases with
increasing x and increases with increasing y, before it returns to near
zero at
x=0, y=0.8 (BLC).
Regarding the fast and slow mass exchange, we suggest that as
hydration affects local structure and, in turn, preferred Co oxidation
states,
a reorganization of La with respect to Ba or Gd may be induced. As
such, local
domains of BLC (ordered or disordered) and BGC or local cation disorder
may be
formed, affecting overall oxidation thermodynamics. The result is that
mass
exchange in varying pH2O
and constant pO2
may be
misinterpreted as only hydration when it could rather be a mix of
hydration and
oxidation. DFT calculations are utilized to establish formation
energies of
protonic defects and oxygen vacancies at favourable hydration sites
surrounding
Gd/La, thermogravimetry is used to determine hydration –
and/or oxidation –
upon isothermal switches between wet and dry atmospheres, and
activation
energies for fast and slow processes are used as indicators of
hydration and
oxidation induced by cation
diffusion.
The research leading to these results has received funding from the
Research Council of Norway (Grant nᵒ 272797 “GoPHy
MiCO”) through the M-ERA.NET
Joint Call 2016.
References
- C. Bernuy-Lopez, K. Høydalsvik, M.-A.
Einarsrud, T. Grande, Materials 9 (2016) (3) 154.
- E.
Vøllestad, M. Schrade, J. Segalini, R. Strandbakke, T.
Norby, J. Mater. Chem. A 5 (2017)
(30) 15743.
- D.S. Tsvetkov, I.L. Ivanov, A.Y.
Zuev, Solid State Ionics 218 (2012) 13.
- R.
Strandbakke, et al., Solid State Ionics
278 (2015) 120.
UM11
Interplay between
electronic structure calculations and advanced characterisation methods
for functional oxides.
Sverre M. Selbach
FACET – Functional Materials and Materials
Chemistry Research Group,
Department of Materials Science and Engineering, NTNU –
Norwegian University of
Science and Technology
Increasing
computational resources together with new and rapidly improving
characterization
techniques expand our possibility to understand structure-property
relationships at the microscopic level, which lies at the heart of
materials
chemistry. In this talk recent efforts to combine DFT calculations with
scanning probe microscopy and synchrotron and neutron total scattering
in the
Functional Materials and Materials Chemistry (FACET) research group at
NTNU will
be presented. The combined theoretical and experimental approach is
illustrated
by the following examples: i) structural disorder across an improper
ferroelectric phase transition, ii) structural disorder in relaxor
piezoelectrics with complex compositions, iii) point defects at
ferroelectric
domain walls with unusual electronic properties. Finally, the
phenomenon of
chemical expansion, imperative to multiple fields of solid state
chemistry, is
revisited, emphasizing the important role of electronic structure.
UM12
Material screening
for battery application – DFT study
P.
Vajeeston, F. Bianchini, H. Fjellvåg
Centre for
Materials Science and Nanotechnology, Department of Chemistry,
University of
Oslo, P.O. Box 1033, N-0315 Oslo, Norway
The demand for novel storage technology for electrochemical
energy is rapidly increasing due to the proliferation of renewable
energy
sources and the emerging markets of grid-scale battery applications.
This
ongoing research for novel battery technology has been considerably
adjuvated
by the emergence of computational first-principle modelling techniques,
such as
Density Functional Theory (DFT), capable of providing accurate insights
into
the performance of functional materials. In our research, we address
two of the
main challenges in the field: the development of efficient electrode
materials
for Li/Na-ion batteries and the research of novel super-ionic
conductors for
application as solid electrolytes. We provide several examples to
illustrate
the usage of DFT to assess the stability of certain phases and to model
the
discharge curve (and thus the cell voltage) of cathode materials, as
well as to
model the ionic diffusivity of alkaline ions in solid electrolytes. The
so
evaluated diffusion barrier is employed, together with geometrical
considerations regarding the dimensionality of the diffusive framework,
to
evaluate the suitability of a set of candidates for efficient battery
applications.
UM13
Tailoring the domain structure in improper
ferroelectrics by
defect chemistry.
D. R.
Småbråten*, T. S. Holstad, D. Meier,
and S. M. Selbach
Department of Materials Science and Engineering, NTNU
Norwegian University of Science and Technology, NO-7491 Trondheim,
Norway.
*E-mail: didrik.r.smabraten@ntnu.no
Understanding the domain wall (DW) dynamics in ferroelectrics is key to
controlling and fine-tune the domain structure, and hence the
ferroelectric properties. The DW dynamics strongly couples to the
defect chemistry, where point defects may act as possible pinning
centers. The overall aim of this study is to give chemical guidelines
for how to control the DW mobility in ferroelectrics by controlling the
defect chemistry. As a model system, we have chosen the improper
ferroelectric hexagonal manganites, because of their complex and exotic
domain structure, including features such as stable neutral and charged
head-to-head and tail-to-tail DWs, and topologically protected vortex
cores. In addition, they possess large chemical flexibility, where
donor and acceptor doping of both cation sublattices, as well as both
oxygen deficiency and excess, are observed experimentally. This makes
the hexagonal manganites an ideal model system for studying the
interplay between the defect chemistry and DW mobility in
ferroelectrics. From density functional theory (DFT) calculations, we
determine the microscopic origin for DW pinning by defects, where the
predicted trends are corroborated by piezo force microscopy (PFM)
images on doped single crystals.
UM14
Effect of Crystallographic Orientation on the
Out-of-Plane
and In-Plane Ferroelectric Properties of BaTiO3 Thin
Films.
T.M.
Ræder,*, E. Khomyakova, J. Glaum, M.A.
Einarsrud, T. Grande
Department of Materials Science and Engineering, NTNU
Norwegian University of Science and Technology, Trondheim, Norway
*Corresponding Author: trygve.m.rader@ntnu.no
BaTiO3 is a widely used ferroelectric material
for dielectric applications, and thin films of BaTiO3
are also of interest for optical modulators, ferroelectric memory, as
well as other devices. Parallel plate electrodes (PPEs) have recently
been used to explore the dependence of the ferroelectric properties on
the crystallographic orientation of BaTiO3 thin
films.[1]
However, interdigitated electrodes (IDEs) are also relevant for many
applications. For this geometry, the electric field is predominantly
in-plane, while for PPEs it is out-of-plane. The two geometries are
schematically shown in Figure 1. IDEs have previously been compared to
PPEs on PZT films, where it was found that IDE structures show higher
coercive fields, saturation polarization, and remnant polarization.[2]
Additionally, recent advances in the understanding of IDEs makes it
easy to correct for geometric factors and extract the relevant material
properties.[3]
Figure 1. Schematic
drawing of a) PPE and b) IDE geometry on BaTiO3 thin
films.
In this work, epitaxial BaTiO3 films are made
using aqueous chemical solution deposition on monocrystalline SrTiO3
(STO) substrates. (001), (011), and (111) oriented STO substrates are
used in combination with both PPE and IDE geometries. Due to a mismatch
in thermal expansion coefficient and the high thermal annealing
temperature of the films, in-plane biaxial tensile strain is developed
during cooling of the films, favoring the development of a-domains.
This affects the ferroelectric properties in films with both PPEs and
IDEs, but the effect is different for the two geometries. Moreover,
IDEs are deposited at several angles relative to the substrate
orientation, so that the properties are measured along different
in-plane crystallographic directions. The BaTiO3
films are
characterized by a combination of X-ray diffraction, transmission
electron microscopy, dielectric spectroscopy and ferroelectric
characterization.
References
- T. Hosokura, A. Ando, T. Konoike. RSC Advances 5.118
(2015): 97563-97567.
- N. Chidambaram, et al. IEEE transactions on ultrasonics,
ferroelectrics, and frequency control 60.8, 1564-1571 (2013)
- R. Nigon, T.M. Raeder, P. Muralt, Journal of Applied
Physics 121.20, 204101 (2017)
UM15
Theoretical approach to understand the origin of
BZY grain
boundary resistance.
Tarjei Bondevika, Ole Martin Løvvika,b, Truls Norbya
aCentre for Materials Science and
Nanotechnology,
University of Oslo, Norway
bSINTEF
Materials Physics, Oslo, Norway
*tarjei.bondevik@smn.uio.no
Y-substituted BaZrO3 (BZY) exhibits high grain
interior
proton conductivity1, but grain boundaries (GBs) have large
resistances, commonly attributed to charge carrier depletion in space
charge layers next to positively charged GB cores2,3. The chemical
environment at the GB is substantially different than in bulk. Even in
materials such as BZY, where normally no secondary phases are observed
at the GB, a structural perturbation over several atomic planes
typically occurs next to the GB core. The structural perturbation
affects the electrical conductivity in two ways. First, charged defects
segregate towards the GB core, leading to a depletion of charge
carriers in space charge layers next to the core, hence reducing the
conductivity. Second, the electrical mobility around the GB core may
also differ compared to bulk, possibly reducing the conductivity
further.
In this work, we investigate the GBs with a theoretical approach to
find the origin of the reduced electrical conductivity. A natural
choice of method is density functional theory (DFT) calculations.
However, such calculations can only be performed on a very limited
number of GBs. As the GBs get more complex, the number of atoms needed
to describe them in a periodic supercell increases. With the
computational cost going roughly as the number of atoms cubed, only the
simplest GBs can be described with DFT.
To reduce the computational cost with up to 90 percent, we combine DFT
with machine learning algorithms. Further, we also apply classical
interatomic potential, which has a computational cost several orders of
magnitude lower than DFT. By combining these approaches, we can
investigate numerous BZY grain boundaries. Specifically, we calculate
the segregation energies and electrical mobility around the GB core,
and relate these quantities to experimentally measured GB resistance.
The ultimate goal is to gain a fundamental understanding of the origin
of the grain boundary resistance, and test the space charge model.
Acknowledgement: This
work is
part of the nationally coordinated project Functional OXides for Clean
Energy Technologies (FOXCET, RCN, 228355), with SINTEF, NTNU and UiO as
active partners.
References
- E. Fabbri, D. Pergolesi, and E. Traversa,
“Materials
challenges toward proton-conducting oxide fuel cells: a critical
review,” Chemical Society Reviews, vol. 39, pp.
4355–4369,
2010.
- X. Guo and R. Wasser, “Electrical properties of
the grain
boundaries of oxygen ion conductors: Acceptor-doped zirconia and
ceria,” Progress in Materials Science, vol. 51, pp.
151–210, 2006.
- J. Nowotny, The CRC Handbook of Solid State
Electrochemistry, CRC Press, 1997.
UM16
Cation disordering in tetragonal tungsten bronzes.
S. S.
Aamlid,* S. M. Selbach, and T. Grande
Department of Materials Science and Engineering, NTNU
Norwegian University of Science and Technology, NO-7491 Trondheim,
Norway
*Corresponding Author: solveig.s.aamlid@ntnu.no
Tetragonal tungsten bronzes (TTB) have the general formula A24Al2C4B10O30.
The A1 and A2 cation sites are of similar size, as shown in Figure 1,
and may accommodate similar types of cations such as alkali and
alkaline earth elements. The solid solution Sr5xBa5-x⎕Nb10O30
(SBN, 0.25<x<0.75) is a common example of a ferroelectric
tungsten bronze. SBN is unfilled, meaning that one of the A sites are
vacant and the remaining five are occupied by a mixture of Ba and Sr.
Ba prefers the A2 site while Sr weakly prefers the A1 site. The effect
of thermal history on the Curie temperature has been reported in SBN in
early works [1]. Quenching from high temperatures is inferred to induce
cation disorder and an increase in Curie temperature for SBN. First
principles DFT simulations of the end members in SBN have demonstrated
that several cation configurations are similar in energy, and that the
degree of disorder will depend on both thermal history and on the Sr/Ba
ratio [2].
Figure 1. The tetragonal
tungsten bronze structure, showing the large pentagonal A2 sites and
the smaller square A1 sites.
Here, the effect of cation disorder in SBN tungsten bronzes is further
investigated. Dense pellets of SBN in four compositions were prepared
by solid state synthesis and quenched from high temperatures to freeze
in various degrees of cation disorder. The effect of thermal history
was characterized experimentally by dielectric spectroscopy,
ferroelectric hysteresis measurements, and X-ray diffraction. The
disordering at high quenching temperatures caused an increase in Curie
temperature, ferroelectric hardening, a contraction of the unit cell in
the a parameter, and the vacancy distribution shifting from the A2 to
the A1 site [3].
References
- R. Guo et al., Ferroelectrics, 93, 397-405 (1989).
- G. H. Olsen, S. M. Selbach, T. Grande. Phys Chem Chem Phys,
17, 30343-30351 (2015)
- S. S. Aamlid, S. M. Selbach, T. Grande, to be submitted
UM17
The chemistries of proton ceramic electrochemical
cells,
Truls Norby
Dept.
Chemistry, Univ. Oslo, SMN, FERMiO,
Gaustadalléen 21, NO-0349 Oslo, Norway
Proton
ceramic electrochemical cells comprise proton conducting ceramic
electrolytes
and various metallic or oxidic electrodes depending on conditions and
processes. Their development from discovery towards commercialisation
is
relatively recent and faces many challenges. However, the use of
optimised
materials and their combinations for various uses such as fuel cells,
steam
electrolysers, and natural gas processing has led to remarkable
progress and attention [1-4]. In
Norway this comprises fundamental and
applied studies at UiO and NTNU in collaboration with SINTEF Industry
as well
as materials synthesis by CerPoTech AS and developments in fabrication
and
applications by CoorsTek Membrane Sciences AS (CTMS).
In
this
contribution, we will review the chemistries at play in state-of-the
art
electrolytes (BaZrO3-based perovskites),
reducing side electrodes
(negatrodes, typically Ni-based cermets), and oxidising side electrodes
(positrodes, typically barium and transition metal-based double
perovskites). This
comprises the thermodynamic and kinetic stability of individual phases
and
their interfaces during fabrication and operation as well as the
kinetics of
solid-state transport and heterogeneous gas-solid reactions at surfaces
and
interfaces including electrodes. Some recent developments in
advancement of
electrocatalytic activity of the electrodes will be highlighted,
covering
promotion of mixed proton electron conduction and in
situ exsolution of nanostructures and catalytic nanoparticles.
References
- C. Duan
et al. «Readily processed protonic ceramic fuel
cells with high
performance at low temperatures», Science,
349
(2015) 1321.
- S. Choi et
al. Exceptional power density and stability at intermediate
temperatures in protonic ceramic fuel cells. Nature
Energy, 3
(2018) 202.
- S.H. Morejudo et
al., "Direct conversion of methane to aromatics in a
catalytic co-ionic membrane reactor", Science,
353
[6299] (2016) 563.
- H. Malerød-Fjeld et al., “Thermo-electrochemical
production of compressed hydrogen from methane with near-zero energy
loss”, Nature Energy, 2 [12] (2017) 923.
Acknowledgement:
Much
of the work to be reviewed has to a large
extent been performed by colleagues at Norwegian partner institutions
SINTEF,
NTNU, and CTMS in numerous RCN-funded projects especially in the
ENERGIX and
NANO2021 programmes (e.g. “FOXCET”), as well as
with international partners in the
EU Energy and FCH JU projects “EFFIPRO”,
“ELECTRA”, and “GAMER” and the
EU ERA
and MERA NET projects “Proton” and
“GoPhyMiCo”.