Det
20. Landsmøte i kjemi
Foredrag
- Abstracts
Fellesarrangemnetet står først,
ellers er foredragene nummerert etter faggruppe:
FE
- Fellesarrangement
AN
- Analytisk kjemi
OM
- Organisk, Makromolekyl- og
kolloidkjemi
UN
- Kjemiundervisning
UM
- Uorganisk kjemi og materialkjemi
HI -
Kjemiens historie
KM
- Kvantekjemi og
modellering
MA
- Matkjemi
Postere
Dette dokumentet oppdateres etterhvert som abstractene kommer inn.
FE -
Fellesarrangement
FE1
Beregning
av likevekt og transport med de små systemers metode (SSM)
Signe Kjelstrup
Institutt for kjemi, Norges Teknisk-Naturvitenskapelige
Universitet
Foredraget tar for seg en ny metode til beregning av termodynamiske
data [1-6]. Metoden er sentral for kjemikere og kalles de små
systemers metode (SSM). To velkjente problemstillinger blir presentert
for å illustrere metoden i bruk, adsorpsjon av CO2
på grafitt [6] og dissosiasjon av hydrogen i en
temperaturgradient [5].
 |
Fig.1.
Små system av varierende størrelse i et reservoir
som
holder temperatur og konsentrasjon konstant. |
Ved likevekt mellom gass og adsorbert lag, eller i den kjemiske
reaksjonen, viser jeg hvordan vi kan skaffe opplysninger om kjemisk
potensial, aktivitetskoeffisienter, reaksjonsentalpier etc. fra en
eneste simulering. Størrelsen på liten test-boks
blir
variert og variasjonen i partikkeltall i den lille boksen blir
undersøkt, se Figur 1. Her ligger all informasjon om
systemet
ved gitte betingelser. SSM bygger på Hills metode for
beregning
av egenskaper til små system. Egenskapene til små
og store
system knyttes sammen med skaleringslover.
Foredraget gir oversikt over diffusjonskonstanter som nylig er beregnet
med metoden [2,4]. Beregningene stemmer svært godt overens
med
eksperimentelle resultater. Vi skal også se at
transportegenskapene til en blanding påvirkes sterkt
når
komponentene deltar i en kjemisk reaksjon [5]. Irreversibel
termodynamikk teori predikerer at en hittil ubrukt transportparameter,
overført varme, forandrer blandingens varme- og masse
overføringsegenskaper. Disse resultatene blir bekreftet ved
simuleringer. Metoden [1-6] kan være nyttige for bedre
modellering av katalyse, membrantransport eller adsorpsjonsprosesser.
Metoden SSM er til implementering i LAMMPS og vil bli åpent
tilgjengelig i 2015.
Referanser
- Sondre K. Schnell, Thijs J.H. Vlugt, Jean-Marc Simon, Dick
Bedeaux, Signe Kjelstrup, Chem. Phys. Letters 504 (2011) 199
- Xin Liu, Sondre Schnell, Jean-Marc Simon, Dick Bedeaux,
Signe
Kjelstrup, Andrè Bardow and Thijs J.H. Vlugt, J. Phys.
Chem. 115 (2011)
12921
- Sondre K. Schnell, Thijs J.H. Vlugt, Jean-Marc Simon, Dick
Bedeaux, and Signe Kjelstrup, Molecular Physics 110 (2011) 1069
- Xin Liu, Ana Martín-Calvo, Erin McGarrity,
Sondre K.
Schnell, Sofía Calero, Jean-Marc Simon, Dick Bedeaux, Signe
Kjelstrup, Andre Bardow, and Thijs J.H. Vlugt, Ind. Eng. Chem. Res. 51
(2012) 10247
- Ragnhild Skorpa, Jean-Marc Simon, Dick Bedeaux, S
Kjelstrup,
Phys. Chem. Chem. Phys. 16 (2014)
19681
- Thuat T. Trinh, D. Bedeaux, J.-M Simon, S. Kjelstrup, Chem.
Phys
Letters 612 (2014)
214
FE2
FE3
Fremstilling og bruk av Ugelstadkuler inn mot
diagnostikk, gensekvensering og cancer immunoterapi.
Geir
Fonnum
Life Techologies AS,
Svelleveien 29, 2004 Lillestrøm, Tel
(+47) 22061246
Geir
Fonnum has M.Sc in Organic
Chemistry from the Norwegian University of Science and Technology in
1985, and
a PhD in the field of Organic/Polymer chemistry in 1989 at the same
University.
His
industrial R&D career
started in the Surface and interfacial group at Dyno Industries.
In
1991 he started working with
Ugelstad spheres for chromatography applications in first at Dynochrom
and later
at Pharmacia Biotech.
In
1999 he moved to Dyno Specialty
Polymers which was acquired by Dynal in 2001. Dynal was acquired by
Invitrogen
in 2005, merged to Life Technologies in 2009 and was acquired by Thermo
Fisher
in 2014.
Geir
has been leading the bead chemistry
development at Dynal since 2001. During
the last 15 years Dynal R&D has developed both magnetic and
non-magnetic
beads products for a broad range of applications ranging from In vitro
diagnostics and proteomics to
semiconductor DNA sequencing (Ion torrent) and beads for cancer
immunotherapy. Dynal
is at the moment the world leading supplier of magnetic beads for these
applications.
In
his talk Geir will show how
Dynal uses the Ugelstad technology to produce magnetic beads for
bioseparations. He will also show how Dynal uses different bead
architectures
to solve the requirements of different biological applications and also
the
results of the major cancer immunotherapy work.
AN -
Analytisk kjemi
AN1
Beyond
Chromatography: Real-time measurements of volatile organic compounds by
PTR-MS
Armin Wisthaler
Kjemisk Institutt, Universitetet i Oslo
Chromatographic separation combined with mass spectrometric analysis
(GC-MS, LC-MS) is usually used for speciation and quantification of
volatile organic compounds (VOCs) in air. Chromatography is, however,
time and labor intensive and off-line analysis does not give real-time
analytical insights. I will herein demonstrate that, for a variety of
analytes and matrices, direct sample introduction followed by chemical
ionization mass spectrometric analysis, is a viable method for
monitoring of VOCs in real-time.
Proton-Transfer-Reaction Mass Spectrometry (PTR-MS) (Fig. 1) combines
the principles of
i) soft chemical ionization of VOCs (via
protonation from H3O+ reagent ions), and
ii) linear and intrinsically quantitative
analyte ion formation in an ion drift tube.
The method excels in high sensitivity (resulting in ppt level detection
limits) and high measurement frequency (up to 10 Hz).
Since its inception in the mid-1990ies, PTR-MS has become a leading
technology in the on-line VOC analysis, spanning a number of research
fields that include environmental chemistry (Fig. 2), food science, and
life sciences. Selected application examples in these fields will be
given.

Fig.
1: Scheme of the PTR-MS instrument
showing the glow discharge ion
source (left), ion drift tube (center), and mass spectrometer (right) |
Fig. 2: 10-Hz VOC data obtained from a
research
airplane that sampled a forest fire plume (figure courtesy: M.
Müller) |
AN2
New gas
chromatography-mass spectrometry (GC-MS) techniques that may
revolutionize environmental analytical chemistry
Peter
Haglund1, Liz Humston-Fulmer2,
Ulrika Olofsson1, Conny Danielsson1
1 Umeå University, Department of Chemistry, 90187
Umeå, Sweden,
2 Leco Corporation, St. Joseph, MI 49085, USA.
In the modern society, large quantities of chemical substances are
used, and more than 30 000 compounds are estimated to be in daily use
in Europe, of which many will reach environment through e.g. the
municipal sewage treatment plant (STP) effluents. In addition to that,
natural production of biogenic halogenated compounds, e.g. marine
toxins, may increase as a result of climate-induced change (CIC). Many
of these chemicals are hazardous to humans and to the environment.
Often these anthropogenic compounds and marine toxins are present at
trace levels together with much more abundant biogenic compounds. Thus,
there is a need for tools to characterize, identify and quantify legacy
and emerging contaminants present in this “chemical
soup”.
Over the last decades, hyphenated techniques combining a
chromate-graphic separation and mass spectrometry have proven to be
well suited for such tasks. In this presentation we will focus on new
and upcoming GC-MS techniques.
The separation power of conventional capillary GC is already quite good
and it may be possible to accommodate several hundreds of peaks in a
one-hour chromatogram. When combined with MS even more compounds can be
resolved. By adding a second GC dimension with orthogonal selectivity
the peak capacity will improve drastically. Using comprehensive
two-dimensional GC (GCxGC) up to 10,000 compounds may be physically
separated. When combined with rapid time-of-flight (TOF) MS and mass
and spectral deconvolution algorithms it is possible to separate and
characterize a significant share of GC amenable compounds present in
complex environmental samples.
Identification may however still be a problem. If the sample
constituents are not present in any of the large electron ionization
(EI) MS libraries, or if there are several compounds with similar
spectra, identification is not possible. In such cases GCxGC
high-resolution TOF-MS can provide valuable complementary information
on the element composition of molecular and fragment ions, which can be
used in manual or in-silico structure elucidation.
In cases when EI do not produce molecular ions identification attempts
are likely to fail and soft ionization techniques would be required.
Traditional chemical ionization (CI) and atmospheric pressure CI often
produce information of psedo-molecular ions, but it is sometimes
difficult to know if this ion is protonated or not. Recently developed
approaches for “soft-EI” may overcome this problem.
Currently, one of the biggest problem is to get sufficiently good
software to efficiently process and quarry the data produced by the
state-of-the-art hardware. Significant improvements are needed in the
peak picking, peak/spectral deconvolution and peak integration
algorithms. It is also room for improvement in the tools for sample
comparison and identification of compounds of interest. Both the
functionality and degree of automation can be improved.
AN3
Barents
Biocentre Lab - Økt verdiskapning fra bioprospektering
Terje Vasskog
Norut
Barents Biocentre Lab (BB Lab) er en bioinkubator i forskningsparken i
Tromsø. Her kan både bedrifter, akademia og
forskningsinstitutter få tilgang til laboratoriearealer og
avansert instrumentering.
Det er en stor satsing på bioprospektering i
Tromsø,
både innenfor næringslivet og i ulike
forskningsgrupper
på UiT - Norges Arktiske Universitet. På BB Lab har
de alle
en mulighet til å benytte instrumentering de ikke ellers har
tilgang til, møtes og diskutere idéer og utvikle
samarbeidsprosjekter.
Per i dag har BB Lab samarbeid med en rekke sentrale aktører
innen bioprospekteringen i Tromsø. MabCent (UiT) har lang
erfaring innen den tradisjonelle marine bioprospekteringen. De samler
inn organismer, ekstraherer, isolerer og bioaktivitetstester ulike
forbindelser i stor skala. De isolerte forbindelsene gir ofte grunnlag
for syntese av større mengder av de isolerte forbindelsene,
eller analoger av disse for å videreutvikle forbindelsen.
MabCent
samarbeider per i dag med Norut på BB Lab om et prosjekt der
slike syntetiserte forbindelser renses og karakteriseres før
de
skal testes for bioaktivitet.
Flere bedrifter er også etablert i BB Lab og jobber innenfor
bioprospektering. Av de største er Barentzymes, et
nyetablert
selskap med 15 ansatte som jobber med enzymer fra organismer i arktiske
farvann. Selskapet fokuserer på å utvikle nye
enzymer til
bruk innen industriell bioteknologi.
Også andre mindre bedrifter har etablert seg på BB
Lab, og
selv om hovedaktiviteten ikke er bioprospektering har flere av dem
vært innom dette fagområdet gjennom samarbeid med
ulike
aktører. Et godt eksempel på dette er
D’liver, et
selskap som tilbyr spesialtilpassede forsøk for å
bestemme
leveropptak, distribusjon og metabolisme av ulike forbindelser.
Kompetansen på dette området ble benyttet i et
samarbeidsprosjekt med Marealis og Norut, der Marealis har oppdaget et
marint peptid med bioaktivitet, og D’liver gjorde
forsøk
på mus med dette peptidet for å se på
opptak og
distribusjon, mens Norut analyserte peptidet i ulike prøver
fra
mus ved hjelp av LC-MS.

Andre aktører, som for eksempel Naturtjenester i Nord
gjør også bioprospektering, men i Nord-Norske
bær
istedenfor marine organismer. I et samarbeidsprosjekt med Norut
analyseres bærekstrakter for ulike forbindelser med gunstig
helseeffekt.
AN4
Metabolomics
– utfordringer og muligheter for kromatografi-MS
Einar Jensen
Institutt for farmasi, Universitetet i Tromsø
Metabolomics er en naturlig medspiller til både genomics og
proteomics. Med å kombinere kunnskaper fra alle de ulike
–omics kan nye pathogene patways oppdages. Metabolomics kan
identifisere molekyler som kan brukes både som prognostikse
og
diagnostiske biomarkørker og evt indikere at bruk av
kostbare
legemidler kan seponeres. Ulike deler av en typisk metabolomics
workflow vil bli diskutert og hvor det vil bli lagt særlig
vekt
på hvilke muligheter og utfordringer ulike varianter av
kromatografi-massespektrometri har innen metabolomics. Definisjon av
metabolomics, study design og bioinformatikk vil og bli kort diskutert.
Prosjektet ”ASIB – Advanced Studies of Inflammatory
Bowel
disease and related disorders” vil og bli presentert.
AN5
Oppdagelse og
analyse av en ny enzymaktivitet
Gustav
Vaaje-Kolstad
Norges miljø- og biovitenskapelige universitet
Mikroorganismer som anvender polysakkarider som næringskilde
tar
i bruk en rekke karbohydrataktive enzymer (CAZymes) for å
oppnå en effektiv nedbrytningsprosess. For fire år
siden
oppdaget vi en ny enzymaktivitet som har vist seg å spille en
viktig rolle i nedbrytningsprosessen, spesielt for solubilisering av
uløselige polysakkarider som kitin og cellulose. Den nye
enzymaktiviteten går under navnet «lytisk
polysakkarid
monooxygenase» (LPMO) og innebærer spalting av
glykosidbindinger med en oksidativ mekanisme. LPMOene benytter et
kopperatom til å aktivere molekylært oksygen,
hvilket ved
en ukjent mekanisme medvirker til oksidasjon av C1 eller C4 karbonet i
glykosidbindingen. Dette gir brudd i polysakkaridkjeden og resulterer i
dannelse av oligosakkarider med enten en aldonsyre (ved C1 oksidasjon)
i reduserende ende eller en 4-ketoaldose (ved C4 oksidasjon) i
ikke-reduserende ende. Det unike med LPMOene er at de
utfører
sin reaksjon direkte på overflaten av en
polysakkaridkrystall. I
foredraget vil jeg lede tilhørerne gjennom historien om
hvordan
en slik tilsynelatende åpenbar enzymaktivitet har forblitt
uoppdaget i så mange år, samt hvordan vi
løste de
analytiske utfordringene relatert til separasjon og identifikasjon av
de oksiderte oligosakkaridene.
AN6
Protein-adducts
and DNA-adducts as
sensitive biological parameters for both biological effect studies and
environmental monitoring purposes
Daniela M. Pampanin
IRIS-International Research Institute of Stavanger
Petroleum related activities in Norway are conducted in shallow seas
contiguous to the Norwegian coastlines and offshore in the North Sea.
Monitoring and assessment of water quality is essential to survey the
potential impact for these activities. Expanding oil exploitation to
more vulnerable area, such as the Artic, requires the development of
even more sensitive methods for the overall understanding of the impact
of oil and chemical contamination in the sea. In the last 10 years,
IRIS, in tight collaboration with Norwegian operators (Norsk olje
&
gass, previously known as OLF) and other national research
institutions, has developed and performed monitoring of the impact of
oil related activity in the North Sea.
Polycyclic aromatic hydrocarbons (PAHs) are common substituents found
in crude oil. Since the occurrence and abundance of PAHs in marine
environments represent a risk to aquatic organisms and ultimately to
humans (through fish and shellfish consumption), there is a constant
need for their characterisation and quantification. The monitoring of
PAH presence in the aquatic environment is therefore a world-wide
activity. Since some of these compounds are well known carcinogens and
mutagens, this contaminant class has been generally regarded as high
priority for environmental pollution monitoring. In fact, the European
Union included these pollutants in the list of priority hazardous
substances for surface waters in the Water Framework Directive
2000/60/EC. There are various environmental monitoring methods that may
be used in order to assess risks of PAH contamination and to classify
the environmental quality of ecosystems. Among those, biological effect
monitoring is defined as the exposure and effect assessment by
determining the early adverse alterations (i.e biological markers) that
are partly or fully reversible in selected organisms.
PAH metabolites have high affinity to nucleic acids (DNA) and proteins,
which may result in adduct formation. This has been shown to be the
case for human serum albumin, which is predominantly alkylated at
histidine146 by diol epoxides of fluoranthene and benzo[a]pyrene. It is
highly likely that the same type of mechanism is operating in animals
and fish. The formation of adducts may result in reduced or impaired
function of genes and proteins.
At present, new indicators of PAH pollution (related to oil production)
in fish in the form of expressed proteins affected by these chemicals
in the marine environment (i.e. by formation of PAH-protein adduct(s))
are under development.
The approach that will be presented aims to provide protein markers
that can track the source of PAH contamination in marine environments
through the identification of PAH adducts in various tissue of
biological fluid of marine organisms.
AN7
Kartlegging av
eksponering for Dieseleksos i Norsk arbeidsliv – hva er
dieseleksos og hvordan kan vi måle dette?
Yngvar
Thomassen
Statens
arbeidsmiljøinstitutt (STAMI)
AN8
Korleis kan vi vite om sjømaten er trygg? Risikovurdering, biologisk variasjon og måleusikkerheit
Amund
Måge, Arne Duinker, Sylvia
Frantzen, Helge Hove, Kåre Julshamn, Tanja
Kögel, Bente Nilsen og Stig Valdersnes
Avdeling
Trygg og sunn sjømat, Nasjonalt institutt for
ernærings-
og sjømatforskning (NIFES), Postboks 2029, 5817 Bergen
Norge er verdas nest største eksportør av sjømat
etter Kina og eksporterte rundt 2,3 millionar tonn sjømat til
ein verdi av drøye 61 milliardar norske kroner i 2013. Det er
også eit relativt stort innalands forbruk av sjømat
både frå handel og frå sjølvfiska mat gjennom
fritidsfiske.
At sjømaten skal vera trygg er eit sjølvsagt krav og
kravet til dokumentasjon har auka jamt sidan 1990-talet. I dette
innlegget presenterer vi det totale overvakingsregimet NIFES har vore
med å byggje opp saman med blant anna Mattilsynet,
Havforskingsinstituttet, FHF-fondet og Nærings– og
Fiskeridepartementet. Grunnlaget for programmet ligg i
prøvetakingsplanar, prøvetaking, prøvebehandling,
analyse og risikovurdering.
For oppdrettsfisk som utgjer rundt ein tredel av norsk sjømat i
volum er prøvetaking og analyse i stor grad underlagt felles
regelverk for all animalsk matproduksjon. Minimums
prøvetakingsregime er ein prøve per 100 tonn
fisk/kjøtt lagt i lovverk (EU 96/23). Her inngår
også krav til analysemetodar og rapportering.
For villfisk fins det ikkje noko tilsvarande regelverk. Samstundes er
det mykje større grad av variasjon i den villfanga
sjømaten. Blant anna er talet på artar som vert fangsta
mykje større enn talet på artar oppdrettsfisk, over 50
norske artar er i sal og endå fleire vert fangsta til
sjølvbruk. Det er også mykje større geografisk
variasjon og større variasjon i størrelse på
fisken.
For villfisken har NIFES valt å satsa på ein kombinasjon av
stikkprøvebasert prøvetaking av få prøvar
for enkelte artar, med ein mykje meir omfattande prøvetaking og
analyse for dei artane som vi har vurdert som heilt sentrale eller som
har hatt ein spesiell risiko for å kome over grenseverdi. Difor
har vi til no utført seks omfattande basisundersøkingar
på til saman fem artar. Dette gjeld torsk, sei, makrell, sild (to
ulike stammer) og blåkveite. Her vert prøvetaking fordelt
over ulike sesongar og i store delar av utbreiingsområda samt at
ein prøver å få med spreiing i størrelse.
Analysane som vert prioritert er for miljøgifter der det er
etablert grenseverdiar i form av øvre grenser eller der vi trur
det kan kome grenseverdiar. Dette har for sjømat vore for
metalla kvikksølv, kadmium, bly, tinn og arsen samt for
dei organiske miljøgiftene dioksin og dioksinliknande PCB,
ikkje-dioksinliknande PCB, bromerte flammehemmarar (PBDE) og
polyaromatiske hydrokarbon (PAH).
Ein del sentrale problemstillingar i skjeringspunktet mellom analytisk
kjemi og biologi er: kor god kvantifiseringsgrense (LOQ) må og
bør vi oppnå, kor stor kjemisk usikkerheit må og
kan vi akseptere, kva er ekstra utfordringar i høve til
kjemiske analysar når grenseverdiar vert oppgjeven som sum av
enkeltkomponentar og vi skal summere enkeltmålingar? Det siste
gjeld til dømes for dioksin og PAH. Her vert også bruken
av Upperbound LOQ som skal brukast for summering av dioksin vesentleg.
Dei analytiske data vert diskutert og sett i samanheng med risiko for
å verta eksponert for høge verdiar av miljøgifter
eller risiko for å fangsta sjømat over grenseverdi
… eller for å eta for lite sjømat.
AN9
Erfaringer og
resultater fra 14 år med internasjonale ringtester for
persistente organiske miljøgifter i mat
Nanna Bruun Bremnes
Folkehelseinstituttet, Divisjon for miljømedisin,
Avdeling
for miljøgifter –kilder og risiko, Postboks 4404
Nydalen,
0403 Oslo
Persistente organiske miljøgifter (POPs) som polyklorerte
dibenzo-p-dioksiner (PCDDs/PCDFs) og polyklorerte bisfenyler er
distribuert globalt og praktisk talt i alle deler av miljøet. De
kan utgjøre en vesentlig helserisiko for mennesker og dyr, og
kan også forårsake skadevirkninger på miljøet.
For å begrense mennesker og dyrs eksponering for POPs gjennom
næringsmidler er det i mange land, blant annet i EU og i USA,
krav til overvåkning av disse miljøgiftene i mat og
fôr. Det er derfor behov for laboratorier verden over som kan
bestemme disse miljøgiftene ved lave konsentrasjoner.
Laboratoriene er ofte pålagt å være akkrediterte i
henhold til ISO- standarder og må vise sin kompetanse gjennom
deltakelse i ringtester.
Divisjon for miljømedisin ved Folkehelseinstituttet har siden
år 2000 arrangert årlige internasjonale ringtester for POPs
i ulike matvarer. Vi har på denne måten kunne tilby et
verktøy for kvalitetssikring av analyseresultater, og har
samtidig hatt en unik mulighet til å studere laboratorienes
kompetanseutvikling innen analyse av disse forbindelsene.
Jeg vil her oppsummere resultatene og erfaringene våre fra 14
år som arrangører av ringtester for POPs i
næringsmidler.
AN10
Consequences of using pooled versus individual samples for
designing environmental monitoring sampling strategies.
Sara Danielsson
Naturhistoriska riksmuseet, Stockholm
Choosing an appropriate sampling strategy for chemical analysis within
environmental monitoring includes the important decision of whether to
sample and store individual or pooled samples. This choice impacts on
future analyses from Environmental Specimen Bank samples. A number of
advantages exist to support using either individual or pooled samples
for temporal trend studies. However, it is important to know the total
and analytical variance to be able to design the best sampling
strategy. Statistical power in temporal or spatial studies is
determined by the random/unexplained sample variation. The relationship
between chemical analytical error and other sources of variation, as
well as the cost for collection, preparation of samples and chemical
analysis, will determine the number of individuals in each pool, and
the number of pools that should be analysed to achieve high cost
efficiency and good statistical power.
In this presentation I will show an example, based on realistic
measures of variation from ongoing Swedish monitoring of contaminants
in marine biota, where the above mentioned components have been
considered in order to design a cost-efficient and statistically sound
sampling strategy.
AN11
Assessing competence in the laboratory
Lorens P.
Sibbesen
Training & Consultancy for laboratories, DK
AN12
Analyse av
fosfor i turbide vannprøver – Spesifikasjon av
krav til analysemetode
Anne Falk
Øgaard og Eva Skarbøvik
Bioforsk Jord og miljø, Ås
Bioforsk rekvirerer en stor
mengde vannanalyser for bestemmelse av partikler og
næringsstoffer i vann. Resultatene inngår i
overvåkningsprogram og forskning. Næringsstoffene fosfor og
nitrogen brukes blant annet som støtteparametre for å
vurdere økologisk tilstand i vannforekomster i forbindelse med
gjennomføringen av vannforskriften. Overvåkingsdataene
inngår i lange tidsserier, noe som understreker betydningen av at
en rekvirert analyseparameter blir analysert på samme måte
hver gang. Erfaring har vist at skifte av laboratorium i et
overvåkningsprogram kan gi uventede og betydelige
«hopp» i analysenivåene.
Følgende momenter gir utfordringer ved analyse av vannprøver:
-Nomenklatur. Spesielt
for fosfor finnes det mange fraksjoner og nomenklaturen for de ulike
fraksjonene er ikke entydig definert. Dette har spesielt gitt
misforståelser om prøven skal filtreres før analyse
eller ikke.
-Metodebeskrivelse.
Metodebeskrivelsene kan være vanskelig tilgjengelig, for eksempel
ved referanse til Norsk standard. Det er ofte uklart hva referansen
refererer til. Er det ekstraksjonsmetoden eller målemetoden? Det
blir heller ikke alltid gitt informasjon om modifikasjoner av
analysemetoden.
-Bruk av underleverandører.
Ved bruk av underleverandører kan en vannprøve bli
fraktet mellom laboratorier for utførelse av ulike analyser. Ved
spørsmål angående prøvebehandling
(filtrering, splitting av prøven, temperatur under oppbevaring
og frakt) kan det være vanskelig å få svar.
-Svar på spørsmål eller henvendelser fra laboratoriet. Disse kan værevanskelig å forstå for folk med liten erfaring i analytisk kjemi.
-Analyse av parametere som ikke er «hyllevare».
Det kan være vanskelig å få analysert en
ønsket fosforparameter, fordi denne ikke inngår i de
kommersielle laboratorienes analyseprogram.
-Mangelfulle metoder.
Analysemetodene som brukes er utviklet for andre typer vann enn turbide
vannprøver og gir dermed ikke sikre svar for denne type
vannprøver.
AN13
Måleusikkerhet
ved sum i multikomponentanalyser
Anders
Torjuul Halvorsen
Laboratorium for klinisk biokjemi, Haukeland
universitetssjukehus
Måleusikkerhet er en viktig del av et analyseresultat, og en
essensiell parameter for å avgjøre en metodes egnethet.
Moderne analyseteknikker er ofte meget spesifikke, og det er flere
vanlige rutineanalyser hvor enkeltkomponenter blir summert til et
analysesvar. Foredraget vil redegjøre for den prinsipielle
mellom en analyse hvor flere komponenter blir målt med én,
uspesifikk måling i forhold til en analyse hvor enkeltkomponenter
blir bestemt og summert. Videre vil estimering og rapportering av
måleusikkerhet i sum av multikomponenter bli gjennomgått,
samt diskusjon rundt praktiske problemer rundt angivelse av
måleusikkerhet for en multikomponentanalyse.
AN14
How to decide
if a method is fit for intended use: The Fitness for Purpose of
Analytical Methods: A Laboratory Guide to
Method Validation and Related Topics
Lorens
P.
Sibbesen
Training & Consultancy for laboratories, DK
OM -
Organisk, Makromolekyl- og
kolloidkjemi
OM1
DNA-Programmed Assembly of
Molecules and Materials
Kurt
V. Gothelf
Center for DNA Nanotechnology
(CDNA), iNANO and Department of
Chemistry, Aarhus University, 8000 Aarhus C, Denmark.
The idea behind our research is to use DNA as a programmable tool for
directing the self-assembly of molecules and materials. The unique
specificity of DNA interactions and our ability to synthesize
artificial functionalized DNA sequences makes it the ideal material for
controlling self-assembly and chemical reactions of components attached
to DNA sequences. We have applied these concepts to assemble and
covalently couple conjugated organic molecules [1] and dendrimers [2].
Recently, we extended this to DNA templated conjugation of DNA to
proteins. In our studies of DNA origami we developed a method to image
chemical reactions with single molecule resolution[3] and to make a 3D
DNA origami box with a controllable lid [4]. More recently, we prepared
a DNA-phenylene vinylene polymer and assembled it on DNA origami for
studies of electronic and optical properties. In extension of this a
method for self-assembly of DNA origami and single stranded tile
structures at room temperature will also be presented [5].

Schematic illustration and AFM image of poly(DNA-phenylene vinylene) on
DNA origami.
References
- Ravnsbæk; J. B et
al. Angew.
Chem. Int. Ed. 2011,
50, 10851–10854.
- Liu, H. et al. J.
Am. Chem. Soc. 2010,
132, 18054-18056.
- Voigt, N. V. et al. Nature
Nanotech. 2010,
5, 200-205.
- Andersen, E. S. et al. Nature
2009,
459, 73-76.
- Zhang, Z. et al. Angew.
Chem. Int. Ed. 2013,
52, 9219
OM2
A tissue-like multifunctional
3D scaffold - Design and
functionalization of resorbable matrices to adapt cell-material
interactions
Anna
Finne-Wistrand
Department of Fibre and
Polymer Technology, School of
Chemical
Science and Engineering, KTH Royal Institute of Technology, SE-100 44,
Stockholm, Sweden
The trend within tissue-engineering research is to obtain material
which stimulate, optimize and control cell-material interactions.
Materials that are bioactive, biomimetic, multifunctional, and
degradable ought to be designed to specifically stimulate cells and
biological processes in a spatial and temporal controlled manner. There
are many parameters to play with, for example surface topography,
hydrophilicity, the mechanical properties, scaffold design and material
functionality. The number of parameters in combination with number of
applications makes it important to develop a tool box for tissue
regeneration, which on one hand fulfils basic requirements and on the
other hand is freely combinable with what is needed in the respective
clinical situation. We are developing this tool box by establishing
synthesis methods and fabrication techniques.
- Synthetic biodegradable
polymers are widely applied today
as 3D
scaffold materials in tissue engineering research because their
chemical, physical and mechanical properties are predictable and
extremely adjustable through variation of monomers, co-polymers, blends
and architectures. However, a new generation of biocompatible and
degradable synthetic polymers with various physicochemical, functional
and biological properties is required to obtain scaffolds which
facilitate advanced tissue engineering. We have functionalized
degradable aliphatic polyesters to extend their properties and the
utility of this class of materials in biomedical applications. For
example, functionalized polyesters from radical ring-opening
polymerizations of ketene acetals and electroactive copolymers.[1-4]
- A variety of fabrication
processes to obtain scaffolds with
highly porous and well interconnected pore structure have been
developed; each combination of material and process has unique matrix
architecture and mechanical properties. To overcome limitations such as
manual intervention, use of toxic organic solvents and use of porogens
and shape limitations, solid free form fabrication was introduced and
fused deposition modeling (FDM) has successfully been used. An
additional advantage is that the scaffolds are with this process built
using layer-by-layer, raising the possibility for hierarchical design.
We have used different fabrication processes over the years and it has
been shown that they are biocompatible and stimulate bone regeneration
both in vitro and in vivo.[4-6]
References
- Undin J. et al., J. Polym.
Sci., Part A: Polym.
Chem. 48:4965-4973, 2012
- Undin J. et al.,
Biomacromolecules, 14:2095-2102, 2013
- Guo B. et al.,
Macromolecules 44:5227-5236,
2011
- Guo B. et al., Chem.mat.,
23:4045-4055, 2011
- Dånmark S. et al.,
J. Bioact. Compat. Polym,
25:207-223, 2010
- Xing Z. et al., Tissue
Engineering, 19:1783-1791, 2013
- Sun Y. et al., J. Biomed.
Mater. Res. Part A,10A:2739-2749,
2012
OM3
Advanced Technical Textiles
Klaus
Opwis
Deutsches
Textilforschungszentrum Nord-West, Adlerstr. 1, D-
47798 Krefeld, Germany
opwis@dtnw.de
For thousands of years mankind uses textiles to protect us against the
weather, as well as to keep us warm and dry (clothes, tents). At the
same time, textiles are important for fashion reasons and as interior
materials (curtains, carpets). Textile materials offer a number of
advantages that make them essential for clothes as well as for
technical textiles. Fabrics can be draped in many different forms - if
needed thousands of times. They can be prepared to be flexible as well
as inflexible. They show a certain permeability for air, vapor and
liquids and textiles combine an enormous stability (especially tensile
strength) with comparatively low weight.
Up to the beginning of the 20th century textile materials based either
on animal (e.g. wool, silk) or plant fibers (e.g. cotton, hemp). With
the rise of synthetic fibers new functionalities are available. Besides
traditional textile applications the area of technical textiles was
born. A technical textile is a textile product manufactured for
non-aesthetic purposes, where function is the primary criterion.
Nowadays, technical textiles can be found everywhere and everybody uses
such technical textiles although not everybody notices them. Some
examples are the conveyor belt at the cash desk in the supermarket, the
safety belt, the tire cords and the airbags in cars or the roofs of
modern sport arenas. Further applications can be found in medicine
(e.g., implant materials, wound dressing), building trade
(fiber-reinforced concrete), protective clothing (e.g., bullet-proof
vests, heat protection, flame retardance), geo- and agrotextiles
(reinforcement of slopes, erosion and crop protection). The sector of
technical textiles is large and grows rapidly with a rate of 4 % per
year. Nowadays the most widely technical textile materials are used in
filter clothing, furniture, hygiene medicals and construction material.
Here, we report our latest approaches on new, innovative technical
textiles, e.g., textiles as carrier material for immobilized catalysts,
polyelectroyte-functionalized textiles for the recovery of noble metals
from industrial waste waters or the decontamination of
chromate-polluted soils and new conductive textiles based on conductive
polymers and its application as textile-based heating elements.
OM4
Triangulenium Salts.
Synthesis, optical properties and self-assembly of cationic
π-systems.
Bo W. Laursen
Nano-Science Center &
Department of Chemistry, University of Copenhagen, Universitetsparken
5, 2100 København Ø,
Denmark.
bwl@nano.ku.dk
Triangulenium salts (Figure 1a) are rigid planar carbenium ions of
exceptional chemical stability, and broad structural diversity.[1] The
key steps in the synthesis of various triangulenium ions are all based
on efficient and selective nucleophilic aromatic substitution reactions
driven by the cationic nature of the precursors and products.[2] After
an introduction to the synthesis of the triangulenium salts the talk
will focus on two topics: 1) Aza/oxa triangulenium dyes (ADOTA+ and
DAOTA+) with long fluorescence lifetimes (τ
≈ 20
ns) and their use in bioimaging and detection of protein-protein
interactions.[3] 2) Self-assembly of amphiphilic derivatives of
amino-trioxatriangulenium salts (ATOTA+). In Langmuir and
Langmuir-Blodgett films as well as in bulk the ATOTA+ salts form
closely packed columnar aggregates with a strong tendency to form
bilayers.[4] However, by changing the associated counterions it is
possible to tune the amphiphilic properties of these aggregates and
thus modify the aggregate superstructures. The interaction between
bilayers of the cationic discotics can be tuned from attractive to
repulsive by choice of anion, leading to either multilayer nanorods
(Figure 1b) or to monodisperse single-walled nanotubes (Figure 1c).[5]

Figure 1. (a) Molecular structures. Cryo-TEM of aggregates formed in
aqueous solutions: (b) multi-layer nanorods (c) mono-disperse single
walled nanotubes formed by amphiphilic ATOTA+ salts.
References
- J. Bosson, J. Gouin, J.
Lacour, Chemical Society reviews 2014, 43, 2824-2840.
- a) B. W. Laursen,
F. C. Krebs, Chemistry - A European Journal 2001, 7, 1773-1783; b) B.
W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B.
Christensen, N. Harrit, J Am Chem Soc 1998, 120, 12255-12263.
- a) E. Thyrhaug, T. J.
Sørensen, I. Gryczynski, Z. Gryczynski, B. W. Laursen, J
Phys Chem A 2013, 117, 2160-2168; b) R. M. Rich, D. L. Stankowska, B.
P. Maliwal, T. J. Sørensen, B. W. Laursen, R. R.
Krishnamoorthy, Z. Gryczynski, J. Borejdo, I. Gryczynski, R. Fudala,
Anal Bioanal Chem 2013, 405, 2065-2075; c) T. J. Sørensen,
E. Thyrhaug, M. Szabelski, R. Luchowski, I. Gryczynski, Z. Gryczynski,
B. W. Laursen, Method Appl Fluoresc 2013, 1, 25001.
- a) J. B. Simonsen, K. Kjaer,
P. Howes, K. Norgaard, T. Bjørnholm, N. Harrit, B. W.
Laursen, Langmuir 2009, 25, 3584-3592; b) J. B. Simonsen, F.
Westerlund, D. W. Breiby, N. Harrit, B. W. Laursen, Langmuir 2011, 27,
792-799; c) F. Westerlund, H. T. Lemke, T. Hassenkam, J. B. Simonsen,
B. W. Laursen, Langmuir 2013, 29, 6728-6736.
- D. Shi, C. Schwall, G.
Sfintes, E. Thyrhaug, P. Hammershøj, M. Cárdenas,
J. B. Simonsen, and B. W. Laursen, Chemistry – A European
Journal, 2014, 20, 6853-6856.
OM5
Endogenous-Inspired
Hydrophobic Drug Delivery to Cancers:
LDL-like
Nano Particles Designed to "Put the Drug in the Cancer's Food"
David
Needham with
Pablo Hervella, Barbara Korzeniowska, Amina Arslanagic, Leena Karmi,
Kasper Glud, Anders Madsen, Malou Reedorf, Ida Appel, Jesper
Skøtt, Koji Kinoshita and Elisa Parra-Ortiz.
Professor, Department of
Mechanical Engineering and Material
Science, Duke University, Durham, North Carolina, USA, and Danish
National Research Foundation Niels Bohr, Visiting Professor Center for
Single Particle Science and Engineering (SPSE), Dept. Physics Chemistry
and Pharmacy, University of Southern Denmark, Odense Denmark
needham@sdu.dk and
http://www.sdu.dk/SPSE
This presentation will discuss our new approaches to nanoparticle
therapeutic drug and imaging agent delivery, as especially applied to
hydrophobic drugs for metastatic cancer. We started
by
asking, “How did nature solve its own delivery of hydrophobic
“drugs” problem?” --the answer
being,
“With Lipoprotein particles”. We are therefore
reverse-engineering the LDL as inspiration for anti-cancer drug
delivery. Motivation for this approach includes the fact that
rapidly growing cancer cells have high numbers of LDLRs, (4-100x
greater than on normal cells); numerous malignancies over-express LDLR
(brain, colon, prostate, adrenal, breast, lung, leukemias, and kidney);
and in patients with cancer, their Low Density Lipoprotein (LDL) count
is even known to go down. Furthermore, an abundance of LDLR
is a
prognostic indicator of metastatic potential, and a propensity to store
cholesteryl ester is a sign of the aggressiveness of a
patient’s
cancer. Our choice of drugs focuses on pathway-specific
growth
and metabolic targets in cancer, that are themselves quite hydrophobic,
such as: Tyrosine Kinase of the growth factor receptor –
(Lapatinib), Wnt pathway (Niclosamide), Fatty acid synthase
(Orlistat), and a variety of Androgen Receptor inhibitors.
Thus,
if we could reverse engineer the LDL, could it inspire a new pure-drug,
ligand-targeted, PET-imageable, nanoparticle, especially for metastatic
disease? “Can we put the drug and the imaging agent in the
cancer’s food?” “Can endogenous
uptake
mechanism be used to make cancer cells take up a drug or imageable
nanoparticle as though it was an LDL of essential materials
--cholesterol, phospholipids, and cholesteryl
ester?” But
instead of being nutrients that feed the cell, Pure-Drug Nanoparticle
(PDN) would retard the cells growth, kill it out right, or cause it to
kill itself.
We will report on the progress we have made towards these goals. This
includes: 1) Developing further a new method [1] for making
nanoparticles by rapid solvent injection, exploring physical and
chemical parameters of solvents, test-materials, PET imaging agents and
drugs; Characterization of LDL receptor (LDLR) expression
level
in human mammary epithelial cell lines, involving techniques such as,
Quantitative Polymerase Chain Reaction, Western Blot, Flow cytometry,
Isogenic cell line technology and Live Cell Imaging; 3) Evaluation of
cell cytotoxicity of formulated peptide-targeted drug nanoparticles;
and 4) the use of the micropipet technique to explore and inform the
nanoscale by observation of solvent injection of miscible and
immiscible solvent-solutions, measurements of interfacial tensions, and
quantifying micro-droplet dissolution rates leading to dug
micro-crystallization or -amorphous formation. All this is
building towards an approach to personalized medicine from the
theranostic side, utilizing PET-imagable metabolic indicators, EPR
evaluation, and pure-drug nanoparticle delivery.
Acknowledgements
The new Center for Single Particle Science and Engineering (SPSE)
established under the auspices of the Danish National Research
Foundation’s Niels Bohr Professorship award to Needham and
SDU.
References
- Zhigaltsev, I.V., Nathan
Belliveau, Ismail Hafez, Alex K.
K.
Leung, Jens Huft, Carl Hansen, and Pieter R. Cullis, Bottom-Up Design
and Synthesis of Limit Size Lipid Nanoparticle Systems with Aqueous and
Triglyceride Cores Using Millisecond Microfluidic Mixing. Langmuir,
2012. 28(7): p. 3633-3640.
UN -
Kjemiundervisning
UN1
Kjemikalier og
avfallshåndtering i skolen.
Brit Skaugrud
Skolelaboratoriet, Kjemisk institutt, Univ. i Oslo
Farlige
kjemikalier brukes både i grunnskolen og i
videregående skole, i kjemi undervisningen i naturfag og i
programfag.
Nettstedet Kjemikalier
i skolen
som er en veileder skrevet spesielt for lærere, skal gi
kunnskap om regelverket
som gjelder for håndtering og oppbevaring av farlige
kjemikalier som brukes i
skolen og være et verktøy for å
få orden på kjemikaliene. Men hva skjer med
kjemikaliene etter bruk? Hva produseres egentlig av farlig avfall
på skolene og
hvordan skal dette avfallet håndteres? Hva må
lærere vite om regelverket
knyttet til farlig avfall og hva trenger de av verktøy for
å håndtere farlig
avfall på en trygg måte? Foredraget skal gi svar
på disse spørsmålene.
UN2
Organisk kjemi -
hvordan få elever interessert i faget.
Yngve
Stenstrøm
Institutt for
kjemi, bioteknologi og
matvitenskap, NMBU, Ås
Realfagsatsingen som myndighetene har satt i gang over mange
år
er vel kjent for de fleste. I praksis går jo denne ut
på at
man skal få ungdom interessert i realfag på tidlige
alderstrinn slik at man får mange til å ta
utdannelse innen
disse fagene. Hvor vellykket dette har vært i praksis kan nok
diskuteres, men det som er helt sikkert er at dersom man ikke
får
barn og ungdom interessert i dette på et tidlig tidspunkt,
så er de tapt med tanke på et universitets- eller
høyskolestudier. Derfor er det viktig ikke bare å
vekke
nysgjerrighet og interesse så tidlig som mulig, men
også
å holde på denne interessen gjennom oppveksten til
en
ferdig utdannet realfagskandidat. Så hvordan kan man
gjøre
dette?
Noe fasitsvar finnes neppe. Og metodene vil selvsagt også
variere
avhengig av fagområdet. Men av egen erfaring både i
undervisning av studenter og ved mange skolebesøk og
besøk ved utstillinger, så er det ingen tvil om at
såkalte knallforelesninger vekker interesse for kjemi.
Imidlertid
er det lett å gå i den fellen at man bare lager
show med
smell, lys og farger, men uten å knytte dette til annet enn
at
”dette er kjemi”. Da er det nok populært
der og da,
men vil også være fort glemt. Derfor er det viktig
å
knytte forsøkene til noe gjenkjennbart og hverdagslig. Og
helst
med en mer eller mindre popularisert forklaring (det vil selvsagt
avhenge av årstrinnet og nivået på
tilhørerne). Kan man i tillegg lage en liten historie rundt
dette, så er det også lettere å huske og
å
kjenne igjen historien og kjemien bak dette. Som kjemiker er derfor
utfordringen å identifisere
”kjemikaliene” rundt seg,
og da definert i videste forstand. Eksempler på slike
kjemikalier
er fett, sukker, proteiner, såper, diverse matvarer generelt,
plastmaterialer, treverk etc. etc. I praksis er det jo absolutt alt vi
omgir oss med. Ved å ta utgangspunkt i slike ting kan man
dessuten ta bort en del myter om at ”alle kjemikalier er
farlige”. I tillegg kan man fortelle at mange av
forsøkene
med disse hverdagskjemikaliene kan man faktisk selv utføre
hjemme om man vil. Så om man gir en liten oppskrift
på hva man kan gjøre, vil det være enda
bedre.
I foredraget vil jeg fokusere på slike eksempler og
også gi noen få eksempler på dette.
UN3
Kan
hydrogen bære energi?
Per Odd Eggen
Skolelaboratoriet, NTNU, Trondheim
Hva
betyr læreplanmålene
“….elevene skal kunne forklare
rollen
til hydrogen som energibærer i fotosyntese og
celleånding”, og ”….
gjøre rede
for struktur og egenskaper til … ATP?” Dette
temaet kom inn i læreplanen i 2006 og innlegget vil
drøfte
utfordringer og muligheter som ligger i disse
læreplanmålene. Er det mulig å se
dem i sammenheng med resten av kjemifaget og på tvers av
faggrensene?
Biokjemidelen
av pensum kan være til nytte på minst to
måter:
Den kan fungere som spesifikke eksempler på tema som er
gjennomgått i Kjemi 1
og Kjemi 2 og dermed fungere som en slags repetisjon.
Virkemåten til ATP kan i hovedsak gå ut
på å drøfte bindinger,
bindingsenergier og termodynamikk.
Når
hydrogen virker som energibærer, dreier det seg om spontane
og
ikkespontane redoksreaksjoner der ”endepunktene” er
spalting av vann og
dannelse av vann. Reaksjonene i elektrontransportkjedene kan virke
svært
kompliserte, men de kan også være egnet til
å repetere grunnleggende
prinsipper.
For
elever som tar biologi i tillegg til kjemi, kan de nevnte
læreplanmålene gi en dypere forståelse av
de kjemiske prosessene i fotosyntese
og celleånding. Dette gjelder egentlig alle elevene, siden
fotosyntese,
celleånding og ATP også er tema i naturfaget.
UN4
Nanopartikler
Ola Nilsen
Kjemisk institutt, Univ. i OSlo
Nanoteknologi
skal være vår mirakelkur for alt. Hva er egentlig
nanoteknologi, og hvorfor fungerer ting annerledes når det
blir
smått?
Naturen er en overlegen produsent av nanomaterialer, men likevel ropes
det varsko når produksjonen bringes inn i en labb. Hva er det
som
gjør oss usikre på hvor dette kan ende?
UM -
Uorganisk kjemi og
materialvitenkskap
UM1
COATINGS
FOR ANTI-ICING APPLICATIONS
Hilde Lea Lein1, Aase
Marie Halvorsen1, Ellen-Krisin Raasok1, Per
Stenstad1, Christian Simon2, Sidsel
Meli Hanetho2
1
Dept. of Materials Science & Engineering, NTNU
2 SINTEF Materials & Chemistry
Ice
accumulation on ships,
offshore constructions and telecommunication equipment is a challenge
when exposed to weather conditions where ice can be formed. Currently,
electrical heaters, hot air sources and addition of chemicals (e.g salt
and glycols), are the most used anti-icing or de-icing techniques.
However, these techniques are not optimal. As alternative, passive
techniques such as material choices, structural surfaces and coatings
have received increased attention.
Hydrophobic
anti-icing
coatings, that successfully repel water and prevent ice accumulation,
are of great importance. This will greatly enhance operational
efficiency, life-time, and safety of constructions and materials
exposed to harsh environment in cold-climate regions.
In
order to increase the
hydrophobicity and anti-icing performance of a coating, a low surface
energy (low degree of wetting) and a characteristic topology of the
exposed surface are of particular interest.
Here
we present a study on
silane-coatings for anti-icing purposes. Different silane precursors
were used as starting materials due to different hydrophobic groups.
Through a hybrid inorganic-organic sol-gel synthesis, the coatings were
prepared, and coated on Si-wafers of different surface topography.
Areas of importance are:
- The
effect of type of fluorosilane precursor
- The
effect of different sol synthesis parameters
- Different coating deposition methods
- The impact of surface morphology
- The effect of thicker coatings (several layers)
The
sols were investigated
by NMR, FT-IR, viscosity and pH. The coatings were studied by using
contact angle measurements, AFM, SEM/EDS, profilometer and white light
interferometry, and icing properties of the coatings were finally
studied.
 |
Fig. 1: A coated Si-substrate with high water contact angle and its
microstructure. |
UM2
HYPER-EXPANDED
FeSe-BASED SUPERCONDUCTORS
Kirill Yusenko,
Serena Margadonna
Department
of Chemistry, University of Oslo,
e-mail: kirill.yusenko@smn.uio.no
The
discovery of superconductivity at critical temperatures, Tc, as high as
48 K in iron selenide based materials with formula MxFe2-ySe2
(M = K, Rb, Cs and Tl) has generated considerable excitement [1-5].
High-temperature solid-state synthesis leads to highly defective and
inhomogeneous samples. Instead, low temperature reaction of FeSe with a
solution of alkali metal in liquid ammonia is a viable route to obtain
single-phase materials. Indeed, ammonothermal synthesis has been
employed and different phases with nominal composition MxFe2Se2
(M = Li, Na, Ba, Sr, Ca, Yb and Eu) were isolated [2]. Intercalation
results in a drastic increase of the interlayer spacing, and the
superconducting temperature reaches 46 K in (NH3)xLi@FeSe
[3]. Here we report a detailed in situ PXRD (SNBL/BM01B beam-line at
the ESRF) and SQUID magnetometry (UiO) study of the reaction between Ba
and FeSe in liquid ammonia solutions; crystal structures and magnetic
properties of final and intermediate phases were investigated to
understand the influence of the interlayer spacing on the properties of
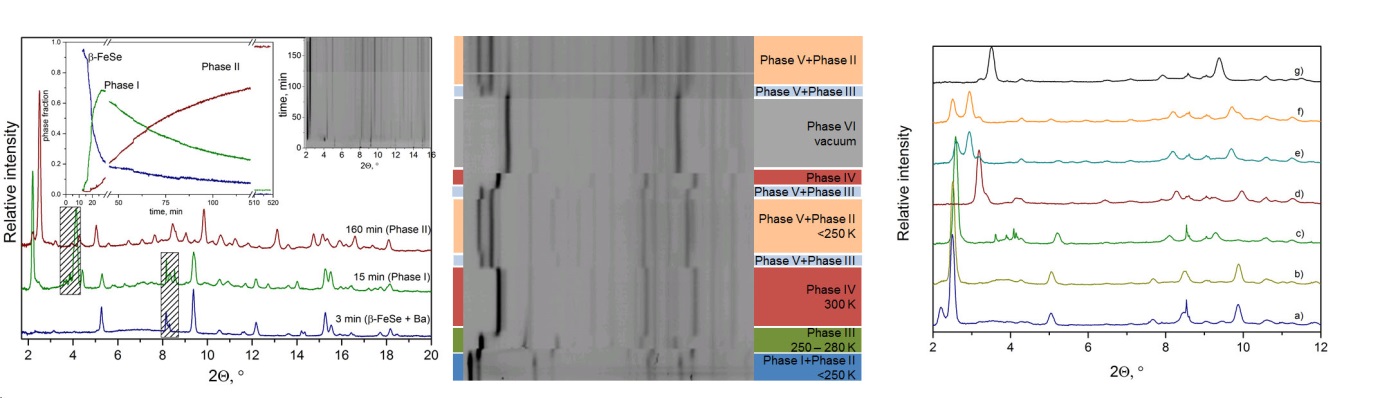
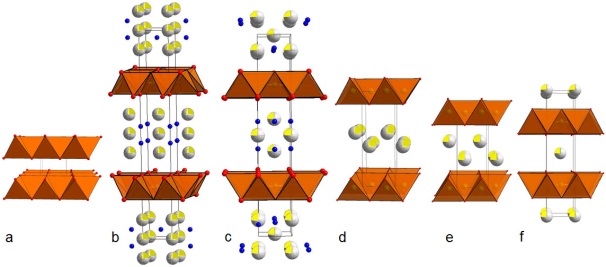
the superconducting and normal state. At least 6 various Ba@FeSe phases
were observed as metastable and stable intermediates between FeSe and
stable ammonia-free Ba@FeSe with critical temperature Tc = 36 K.
Intermediate phases show variations in interlayer distances (d = 13.14
– 8.38 Å, see Figure) and as a result various
superconducting temperatures (Tc = 39 – 34 K) [4-5]. In
particular phase I and phase II show the largest interlayer spacing
ever reported. Reaction FeSe + Ba → Phase I has
τ½ ~
2-3 min and subsequential reaction Phase I → Phase II is
relatively slow ( τ½ ~ 70
min). Importantly,
phases I and II show the largest interlayer spacing ever reported in
any analogous system but not the highest Tc. This observation
contradicts the common understanding of the properties of these
materials and opens a number of questions on the real influence of the
interlayer distance on the superconducting mechanism.
Figure.
Left: PXRD data
(λ = 0.504850 Å) obtained for Ba and
β-FeSe mixture
at 200 K (diffraction lines characteristic for Ba and BaNHx
phases are striked out). Insets show time dependence of diffracted
intensity and phase fraction for β-FeSe, Phase I and Phase II
obtained from Rietveld refinement. Middle: PXRD data for Ba +
β-FeSe after completion of transformation to Phase I. Right:
crystal structure of Ba@FeSe (phase VI).
References
- Tian-Ping
et al. (2013): Chin. Phys. B 22, 087412;
- Li
et al. (2012): Nature Phys. 8,126;
- Wang
et al. (2012): Scientific Rep. 2, 426;
- Yusenko,
Margadonna et al. (2014): ChemComm, submitted;
- Yusenko,
Margadonna et al. (2014): JACS, submitted
UM3
DEVELOPMENT
OF NOVEL BIODEGRADABLE HYBRID NANOPARTICLES FOR CANCER DIAGNOSIS AND
THERAPY
Fuad Karimov1,
Juan Yang1, Nicolas Rival1,
Huaitian Bu1, Stephan Kubowicz1,
Christian Simon1, Tore-Geir
Iversen2
1
Materials and Nanotechnology Sector, SINTEF materials and Chemistry,
Forskingsveien 1, 0314, Oslo, Norway
2 Department of Biochemistry, Institute for Cancer Research The
Norwegian Radium Hospital, Montebello, 0379 Oslo, Norway
Nanoparticles
(NPs) developed from polyhedral oligomeric silsesquioxane (POSS)
structures open new perspective in the field of drug delivery. Due to
their nanocaged structure consisting of an inner inorganic framewrok of
silicon and oxygen atoms, and an outer shell of organic functional
groups, these NPs have a unique biomedical application. By the
implication of organic chemistry and polymer science different types of
POSS based NPs can be obtained.
Nanomicelles
can be formed
by creating an amphiphilic POSS structure, a POSS structure partially
modified with organic fatty acid and long PEG chain. Formation of
nanomicelles can facilitate the drug loading of hydrophobic drugs. This
will enhance the bioavailability of these drugs.
In the actual work polyhedral oligomeric silsesquoixane POSS structure
was modified with behenic acid and PEG methacrylate chains. Different
ratios have been used in order to prepare these NP's. POSS based
structure is expected to form nanomicelles in the aqueous solution and
be stable during long period of time. The results obtained from
particle size measurement show that POSS based nanomicelles form
agglomerates after dialysis while the average size of 100nm was
obtained before dialysis (having organic solvent inside the sample).
Preliminary study of hydrophobic drug loading was tested using NR668 a
hydrophobic dye Nile Red. Stability of nanomicelles in cell medium was
as well investigated.
UM4
Engineering
the morphology, composition and structure of PtRh Nanoparticles by
Microwave Irradiation Synthesis
Maria Kalyva,
Helmer Fjellvåg and Anja Olafsen Sjåstad
Department
of Chemistry, University of Oslo
PtRh
bimetallic
Nanoparticles (NPs) capped with polyvinyl pyrrolidone (PVP) were
synthetized by Microwave Irradiation (MIW) dielectric heating. We were
able to tune the morphology of the NPs acquiring octopod-cubes, cubes,
truncated cubes and small spheres by increasing the molar ratio of PVP
to Pt- and Rh precursors, keeping the microwave irradiation time
constant (5min). The NPs were characterized by High Resolution
Transmission Electron Microscopy (HRTEM), Energy Dispersive X-ray
Spectroscopy (HRTEM-EDS), X-Ray diffraction (XRD) and X-ray
Photoelectron Spectroscopy (XPS). TEM analysis revealed near
monodispersed NP distributions, covering the range from 3 to 18 nm.
XRD, XPS and EDS results show that the produced NPs consist mainly of
Pt, while Rh is detected only for the lowest PVP concentration. XPS
measurements indicate that the surface is enriched in Rh, indicating a
core shell structure. The composition of the PtRh NPs was tuned by
changing the time parameter of the MIW synthesis. Increasing the total
MIW time up to 20 min we were able to prepare alloy Pt100-xRhx
NPs with x < 50, while keeping equimolar precursor’s
concentration, see Fig. 1. Incorporation of Rh in the alloying process
make it complicated to control shape and the size of the NPs.
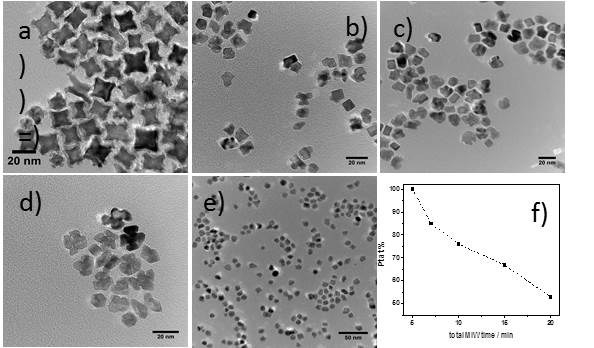
Fig. 1: HRTEM images of PtRh NPs prepared at a)5min, b) 7min, c) 10min,
d)15min and e) 20min of microwave irradiation. Graph f) shows the at.%
of Pt calculated from the EDS spectra.
References
- I.Bilecka
and M. Niederberger, Nanoscale, 2010, 2, 1358-1374.
- L.Dai, Q.Chi, Y. Zhao, H.Liu, Z. Zhou, J. Lii, T.Huang,
Materials Research Bulletin, 2014, 49, 413-419.
- J.Y.Park, Y.Zhang, M. Grass, T. Zhang and G.A. Somorjai,
Nano Lett., 2008,8 (2).
UM5
A
MASTER MODEL FOR PROTON CONDUCTING MATERIALS
Truls Norby,
Sindre Ø. Stub
Department of Chemistry, University of Oslo, FERMiO,
Gaustadalléen 21, NO-0349 Oslo, Norway
Proton
conduction is
observed in materials over temperatures ranging from ambient to above
1000°C, and applications comprise as different materials as the
polymer proton exchange membranes (PEMs) operating below 100°C,
the
solid acids like CsH2PO4
operating around 200°C, and proton conducting oxides, operating
typically at 600°C. The discoveries of new families of proton
conductors are claimed, such as high-temperature N-containing polymers
like poly-benzimidazole, pyrophosphates like SnP2O7,
and nanograined or nanoporous ceramics of otherwise oxide ion
conducting YSZ and GDC. Most of the proton conductors exhibit a maximum
in conductivity with increasing temperature, reflecting the combined
action of increasing mobility of protons or protonated species on the
one hand, and decreasing concentration through dehydration. There has
nevertheless been little general recognition of common features, and
moreover, there are conflicting reports of phase purity, proton
conductivity, and interpretation and mechanisms involved.
In
this contribution, we
present for the first time a more global view - a kind of a master
model and a generic master Arrhenius-type temperature dependency - of
the protonic conductivity contributions that a typical materials sample
may have. For an acceptor-doped oxide ceramic, this involves conduction
of free protons, peaking at typically a few hundred degrees centigrade,
and protonic conduction in the ad- and absorbed water in open grain
boundary cores and porosity. The latter comprises conduction in single
and multiple layers of adsorbed water - increasing with decreasing
temperature - and in condensed water, increasing strongly towards the
dew point in the surrounding atmosphere. Both carrier concentration and
hydration can be affected by "doping" the material and hence internal
surfaces with oxyacid groups.
As
temperature is lowered
further the condensed water exhibits a maximum in conductivity and
decreases with decreasing mobility. Freezing may add to this.
All in all, the temperature dependency of the protonic conductivity of
a ceramic oxide exhibits two maxima at high and ambient temperatures,
respectively, and the latter has many features in common with the
conductivity of proton exchange polymers membranes (PEMs) and
acid-doped pyrophosphates, and we will discuss this in terms of a
uniform model.
Acknowledegement
Work
supported by the Research Council of Norway, RENERGI "NaProCs" 216039.
UM6
DEFECT
CHEMISTRY OF HEXAGONAL MANGANITES FROM FIRST PRINCIPLES
Sandra
Helen Skjærvø1,
Thomas Tybell2, Sverre Magnus Selbach1
1
Dept. Materials Sci. Eng., NTNU
2 Dept. Electronics Telecom., NTNU
Hexagonal
manganites have attracted considerable attention due to their
multiferroic properties [1, 2], but has also recently shown promise for
energy technology purposes. Due to their layered crystal structure, the
hexagonal manganites can accommodate interstitial oxygen, which are not
common in perovskites. Hexagonal Dy1-xYxMnO3+δ can
accommodate
excess oxygen up to δ < 0.35 at relatively low
temperatures of
250-400 ºC [3], opening the possibility for use as oxygen
storage
materials at much lower temperatures than the present state-of-the-art
materials [4, 5]. Oxidation of the multivalent manganese ions could be
the charge compensating mechanism, creating holes in the valence band
of YMnO3 and p-type semiconductivity. [6].
Hexagonal
manganites can
also be oxygen deficient, and show strongly anisotropic chemical
expansion. Flexoelectricity in hexagonal manganite thin films has been
attributed to oxygen vacancies creating stress fields due to the
chemical expansion. [7, 8]. Computational studies on chemical expansion
will become increasingly important as transition metals oxides are
integrated into electronic circuitry. [9].
A-site
deficiency will also
be addressed as our preliminary studies have demonstrated that YMnO3
can accommodate up to 30 % Y deficiency with surprisingly small
structural changes.
Fig.
1: a) Interstitial
oxygen in most stable position in YMnO3. b) Defect formation energy for
interstitial oxygen as a function of chemical potential of oxygen in
YMnO3
References
- Tyson
et al. J. Appl. Phys. 110 (2011) 084116.
- van Aken et al. Nature Mater. 3 (2004) 164.
- Remsen and Dabrowski. Chem. Mater. 23 (2011) 3818.
- Hendriksen et al. Catal. Today 56 (2000) 283.
- Vente et al. J. Solid State Electrochem. 10 (2006) 581
- Kalinin and Spaldin, Science 341 (2013) 858.
- Ren et al. Appl. Phys. Lett. 103 (2013) 152905.
- Aschauer et al. Phys. Rev. B 88 (2013) 054111.
- Adler J. Am. Ceram. Soc. 84 (2001) 2117.
- Kalinin et al. ASC Nano, 6 (2012) 10423.
UM7
HYDRATION
AND INTERCALATION IN RUDDLESDEN-POPPER PHASES
Vegar Øygarden1,
Chris I. Thomas1, Helmer Fjellvåg2
and Anja O. Sjåstad2.
1
Centre for Materials Science and Nanotechnology, University of
Oslo.
2 Department of Chemistry, University of Oslo
Ruddlesden-Popper
(RP) type
oxides, An+1BnO3n+1, possess a wide range of technologically important
properties; i.e. thermoelectricity, colossal magneto-resistance, mixed
conductivity and high temperature superconductivity. RP oxides are also
currently of high interest in solid oxide fuel cell (SOFC) technology.
The RP structure consists of n blocks of perovskite type units (ABO3)
separated by a layer of rock salt type (AO). Certain A4B3O10 (n=3, RP3)
materials are prone to intercalation reactions with water, carbonate
and simple alcohols after treatment in reducing atmospheres [1, 2].
Several show major oxygen intercalation. The intercalated species
locate to the rock salt layers, however, the process and the
accompanying structural and physical changes are poorly understood.
Their electric transport properties are affected by intercalation, such
as changing from mixed to ionic conduction. The properties can be
modified by careful tuning of the oxygen stoichiometry and type and
concentration of the intercalating species. In addition to the focus on
intercalation, we explore novel solid solution systems with intriguing
physical properties, including redox activity and vacancy ordered
superstructures. The goal of this presentation is to demonstrate the
interplay between the structural, electronic and magnetic properties of
RP3 phases as a function of intercalation.

Fig. 1: Structural change throughout
water intercalation and de-intercalation process.
References
- L.
Jantsky et al., Inorg. Chem. 51 (2012) p. 9181-9191.
- M.
Lehtimäki et al., J. Sol. State Chem. 204 (2013) p. 95-101.
UM8
REVISITING
RHOMBOHEDRAL LEAD METANIOBATE: CRYSTAL STRUCTURE AND FUNCTIONAL
PROPERTIES
Gerhard H. Olsen,1
Magnus H. Sørby,2 Bjørn C.
Hauback,2
Sverre M. Selbacħ1 and Tor Grande1
1
Norwegian University of Science and Technology (NTNU), Department of
Materials Science and Engineering, Trondheim
2 Institute for Energy Technology (IFE), Kjeller
Lead
metaniobate (PbNb2O6)
can exist both as a stable rhombohedral and a metastable orthorhombic
tungsten-bronze-type polymorph.[1,2] Although the tungsten-bronze is a
well-known ferroelectric material, the rhombohedral polymorph (Fig. 1)
has been far less studied. The crystal structure and energetic
stability of the stable rhombohedral polymorph of lead metaniobate is
re-examined by powder X-ray diffraction and powder neutron diffraction,
in combination with ab initio calculations.[3] We show that the structure
is described by the polar space group R3, in contradiction to the
previously reported space group R3m. The polar space group opens up for
possible device applications of the material, and investigations of the
high-temperature behaviour of the materials also hint towards
interesting functional properties.
Fig. 1: The crystal structure of rhombohedral
lead metaniobate. Left: Single layer built from Nb2O10
dimers; Right: Stacking sequence of layers with Pb2+
in channels along [001].
References
- G.
Goodman, J. Am. Ceram. Soc. 36 (1953) 368–372.
- M. H. Francombe, Acta Crystallogr. 9 (1956)
683–684.
- G.
H. Olsen et al., Inorg. Chem. (2014) DOI: 10.1021/ic5012378
UM9
CRYSTAL
CHEMISTRY AND THERMAL PROPERTIES OF RARE EARTH BOROHYDRIDES
Christoph Frommen,
Michael Heere, Magnus H. Sørby, Bjørn C. Hauback
Institute
for Energy Technology, P. O. Box 40, NO-2027 Kjeller
Rare earth (RE)
borohydrides have received
considerable attention during the past 5 years due to their rich
crystal chemistry [1-4] and potential as both solid state hydrogen
storage materials and solid state electrolytes [5]. Mechanochemical
synthesis that utilizes a metathesis reaction between a RE-chloride and
an alkali metal borohydride (mostly LiBH4) is
now the
standard technique for the synthesis of RE-borohydrides.
RE-borohydrides form distinct structure types which are determined by
the ionic radius of the RE and its electronic configuration. The early
lanthanides La, Ce, Pr, and Nd form LiRE(BH4)3Cl
compounds (cubic; I-43m), where each RE is octahedrally coordinated by
three BH4- units and three Cl atoms, while the RE and Cl atoms in the
unit cell form a distorted Re4-Cl4
cube. Sm, Gd, Tb, Er and Yb form α-RE(BH4)3
(cubic; Pa-3) with a possible polymorphic transition to β-RE(BH4)3
for Y, Yb (cubic; Pm-3m or Fm-3m). α/β-RE(BH4)3
borohydrides are related to the ReO3-structure
type where the RE atoms are situated at the corners of a cube and the BH4
units lie along the edges leading to an octahedral coordination of RE
by six BH4 units. The smallest lanthanides Yb
and Lu form tetrahedral [RE(BH4)4]-
anionic complexes that are stabilized by Li+ cations (tetragonal;
P-42c) in analogy to LiSc(BH4)4.
Furthermore, Sm and Gd show transitions to the LiRE(BH4)3Cl
structure type that is observed for the largest lanthanide ions [2, 3].
Composite
mixtures between a RE-borohydride and LiBH4 are
interesting for hydrogen storage purposes because excess LiBH4
can be destabilized via the formation of RE-hydrides which results in
significantly reduced desorption temperatures [3]. We have investigated
a wide range of composites based on 6LiBH4-RECl3-3LiH
and present experimental results based on in/ex-situ powder X-ray
diffraction, thermogravimetric and caloric measurements, and cycling
experiments for La- and Er-based borohydrides.
References
- C.
Frommen, N. Aliouane,
S. Deledda, J.E. Fonneløp, H. Grove, K. Lieutenant, I.
Llamas-Jansa, S. Sartori, M.H. Sørby, B.C. Hauback, Journal
of
Alloys and Compounds, 496 (2010) 710-716.
- C. Frommen, M.H. Sørby, P. Ravindran, P.
Vajeeston, H.
Fjellvåg, B.C. Hauback, Journal of Physical Chemistry C, 115
(2011) 23591-23602.
- J.E. Olsen, C. Frommen, T.R. Jensen, M.D. Riktor, M.H.
Sørby, B.C. Hauback, RSC Advances, 4 (2014) 1570-1582.
- J.E. Olsen, C. Frommen, M.H. Sørby, B.C.
Hauback, RSC Advances, 3 (2013) 10764-10774.
- M.B. Ley, S. Boulineau, R. Janot, Y. Filinchuk, T.R.
Jensen, Journal of Physical Chemistry C, 116 (2012) 21267-21276.
UM10
STRUCTURAL
AND MAGNETIC ASPECTS OF La4(Co1-xNix)3O10+δ
(0=< x =<1)
M. U. Nagell,
S. Kumar, M. H. Sørby, H. Fjellvåg, A. O.
Sjåstad
Department
of Chemistry, University of Oslo
La4(Co1−xNix)3O10+δ
bridges two Ruddlesden-Popper RP = 3 phases with quite different
properties: Pauli-paramagnetic La4Ni3O10+δ
with metallic like conductivity [1] and the antiferromagnetic La4Co3O10+δ
semiconductor [2]. La4Ni3O10+δ
is reported as orthorhombic [1] whereas La4Co3O10+δ
is monoclinic [2, 3]. However, a broadening of (117) in the X-ray
diffractograms suggests monoclinic distortions also for La4Ni3O10+δ.
A number of structural deformations (and space groups) as function of
composition (x) are proposed by Amow et al. for La4(Co1−xNix)3O10+δ
[4].
The
current neutron diffraction analysis indicates no structural change
when going from La4Co3O10+δ
to La4Ni3O10+δ.
For all compositions it is feasible to adjust the oxygen content
(δ) by annealing at fixed temperatures by varying oxygen
partial
pressure, tuning the amount of trivalent Co- and Ni-cations. Oxygen
analysis is done by cerimetry and TGA. The magnetic and electric
properties are strongly composition dependent (x,
δ).
Finally, the results from the various measurements is summarised in a
phase diagram of structural and electronic properties of the complete
solid solution La4(Co1−xNix)3O10+δ.
References
- Z.
Zhang, M. Greenblatt, J. Solid State Chem. 117 (1995) 236
- O. H. Hansteen, H. Fjellvåg, J. Solid State Chem.
141 (1998) 212
- H. Fjellvåg, O. H. Hansteen, B. C. Haubach, P.
Fischer, J. Mater. Chem. 10 (2000) 749
- G. Amow, J. Au, I. Davidson, Solid State Ionics 177 (2006)
1837
UM11
ZEG
Power - more energy and less emissions
Bjørg
Andresen, Managing Director
ZEG Power AS, c/o IFE P.O. Box 40, 2027 Kjeller
The
ZEG-technology (Zero Emission Gas - ZEG®)
is a hybrid technology for co-production of electricity and hydrogen
from hydrocarbon fuels, with integrated CO2-capture. The main strengths
of ZEG®
for energy
production are high overall efficiency, in the range of 70 to more than
80% depending on the plant size, and the flexibility; all types of
carbon based fuels can be used, product composition can be varied
dependent on market demand and applications and scale are from small
scale distributed plants based on biogas to industrial scale gas power
plants.
The
basic technologies in
the ZEG-concept are electricity production by SOFC (solid oxide fuel
cells) and hydrogen production by a modified steam reforming reaction,
(SER - sorption enhanced reforming). Close thermal integration of the
two basic technologies is necessary in order to obtain a high total
efficiency.
The
technology will be
presented included main results from the basic technologies, status and
plans for further technology development.
UM12
Steam
to hydrogen using high temperature proton ceramic electrolyser cells
Einar
Vøllestad and Truls Norby
Department of Chemistry and Centre for Materials Science and
Nanotechnology, University of Oslo
Renewable energy sources are gradually becoming more important to the
largest energy markets worldwide, as fossil fuel and nuclear power
plants are phased out. As the market share of intermittent energy
sources such as wind and solar continue to grow, it is of vital
importance to develop cost efficient and clean energy storage
technologies to store peak hour energy. High temperature electrolysis
(HTE) of steam offers high efficiency of conversion of renewable and
peak electricity to H2 and may increase
efficiency further
by utilising available sources of heat and steam from solar,
geothermal, or nuclear power plants. Technologies developed to date
comprise solid oxide electrolyser cells (SOECs) utilising oxide ion
conducting electrolytes operating by virtue of necessity around 800
°C. They produce hydrogen on the steam feed side, and
separation
and drying of H2 costs energy and add plant
complexity and
footprint. In comparison, a high temperature proton conducting
electrolyte will instead pump protons (H+) and form dry H2,
leaving O2
on the steam side. Such proton ceramic electrolyser cells (PCECs) thus
require less separation process stages and can produce pressurised dry
H2 directly. Protons exhibit lower activation energies than oxide ions,
and ceramic proton conductors should be able to operate at lower
temperatures – 600-800°C – i.e., closer to
the ideal
range for integration with solar and geothermal plants.
This
contribution will
outline the general concepts of steam electrolysis with emphasis on the
different electrochemical processes that occur throughout the
electrolysis cell, and the remaining material challenges that must be
resolved for proton ceramic electrolyser cells to be competitive as a
cost efficient energy storage technology: i) Development of
novel H2O-O2
anodes comprised of composite oxide materials that display mixed
proton-electron conductivity, high catalytic activity towards water
splitting, and compatibility with the electrolyte material; ii)
development of scalable fabrication routes for complete electrolysis
cells that can withstand 50 bar steam at 600-800°C for long
durations.
UM13
STRIKING
HYDROGEN UPTAKE BEHAVIOR DIFFERENCES IN CPO-27 MATERIALS INDUCED BY
METAL SUBSTITUTION
Mali H. Rosnes,1
Martin Opitz,1 Wiebke Lohstrohb,2
Jan Peter Embs,3 Peter A. Georgiev,4,5
Pascal D. C. Dietzel1
1
Department of Chemistry, University of Bergen, P.O. Box 7803, N-5020
Bergen, Norway.
2 Heinz Maier-Leibnitz Zentrum (MLZ), Technische Universität
München, Lichtenbergstraße 1, D-85748 Garching,
Germany.
3 Laboratory for Neutron Scattering, Paul Scherrer Institut, CH-5232
Villigen, Switzerland.
4 Department Chemistry, University of Milan, 21 Via Golgi, I-20133
Milan, Italy.
5 Faculty of Chemistry and Pharmacy, University of Sofia, James
Bourchier 1, 1164 Sofia, Bulgaria
Metal-organic
framework
materials have shown remarkable hydrogen uptake and sorption
properties. Among the most intriguing series of compounds are the
isostructural microporous coordination polymers [M2(dhtp)]
(CPO-27-M, M-MOF-74 or M2(dobdc)),
in which a high concentration of coordinatively unsaturated metal sites
results in high initial heats of adsorption.[1]
We
present a comparative
study of hydrogen gas adsorption experiments on CPO-27-Cu and -Mn,
which show striking differences in their hydrogen uptake behaviors
which can be attributed to the difference in interaction between
hydrogen and the respective metal cation incorporated in the framework
structure. Inelastic neutron scattering experiments were carried out to
confirm the remarkable difference and gain further insight into the
adsorption process. Firstly, CPO-27-Cu demonstrates the lowest
isosteric heat of adsorption for H2 of all the CPO-27-M materials
reported to date, where M = Ni, Co, Mg, Zn, Mn, and Fe,[1] and
CPO-27-Mn the second lowest. Secondly, all the reported CPO-27
materials show two steps in the adsorption isotherm and two distinct
values corresponding to the first and second adsorption sites in the
heats of adsorption, which are not observed for the CPO-27-Cu. Thus,
the open metal site and second adsorption site are energetically
equivalent in CPO-27-Cu, and there is no preference for the hydrogen
gas at the open metal center.
References
- P.
D. C. Dietzel et al. Chem. Commun. 2010, 46, 4962-4964
UM14
SODIUM-
AND POTASSIUM CONTAINING THIN FILMS BY ATOMIC LAYER DEPOSITION
Henrik H.
Sønsteby, Øystein S.
Fjellvåg, Ola Nilsen and Helmer Fjellvåg
Department of
Chemistry, University of Oslo
Sodium-
and potassium
containing complex oxides exhibit a range of properties important for
applications in present and future materials technology. Examples of
such compounds include ferroelectric KxNa1-xNbO3
[1], the battery cathode material NaxMnO2[2]
and thermoelectric NaxCoO2[3],
finding their use on devices such as sensors, actuators and devices
with need for integrated energy supply. As devices become smaller, the
need for techniques enabling deposition of thin films with
sub-nanometer thickness control and high conformality becomes
increasingly important. Atomic layer deposition is a well-established
technique that accommodates these criteria[4]. Processes for depositing
oxides of most common elements have already been reported[5], with
exceptions including many of the important alkali metals due to lack of
suitable precursors. In this research, we highlight some crucial
challenges of atomic layer deposition of compounds containing sodium
and potassium and how these can be overcome, including presentation of
suitable precursors. Deposition of some technologically interesting
materials is used to support the impact this progress may have for
future thin film research.

Fig. 1: The two precursors enabling atomic
layer deposition of sodium- and potassium containing thin films.
References
- Nakashima
et al., Jpn. J. App. Phys. 46, L311, (2007)
- Doeff et al., J. Electrochem. Soc. 141, 11, L145-L147 (1944)
- Lee et al., Nat. Mater. 5, 537-540, (2006)
- George et al., Chem. Rev., 110, 111-131, (2010)
- Miikkulainen et al., J. Appl. Phys., 113, 021301, (2013)
UM15
LUMINESCENT
LANTHANIDE TITANATES BY ALD
Per-Anders
Hansen1, Helmer Fjellvåg1, Terje
Finstad2, Ola Nilsen1
1
Department of Chemistry, University of Oslo
2 Department of Physics, University of Oslo
LUMINESCENT LANTHANIDE TITANATES
AND LANTHANIDE YTTERBIUM TITANATES HAVE BEEN GROWN BY ALD FOR LIGHT
CONVERSION
Lanthanide oxides is an important class of functional materials, used
as light converters in phosphor materials, active media in lasers, fuel
cells and magnetic materials to name a few. Atomic layer deposition
(ALD) gives unique possibilities in tailoring doping profiles and
compositions not easily reproduced by other methods. Controlling the
film growth and growth parameters of the lanthanide oxides thus enables
novel structures and materials to be synthesized.
Based on our previous work on ALD films of Ln2O3[1],
EuxTiyOz[2]
and nanolaminates of Eu2O3/TiO2
sandwich structures[3], we here explored the luminescent properties of
LnxTiyOz
and (Ln,Yb)xTiyOz
film materials. The goal is to achieve efficient UV-to-NIR light
conversion through UV absorption in TiO2 and
1000 nm emission of Yb3+, with or without and
intermediate step through a second lanthanide Ln3+.
Fig. 1:
Visible and NIR emission of LnxTiyOz
thin films, excited by 325 nm HeCd laser.
References
- P.-A.
Hansen, H. Fjellvag, T. Finstad and O. Nilsen, Dalton Transactions,
2013, 42 (30), 10778 - 10785.
- P.-A. Hansen, H. Fjellvåg, T. G. Finstad and O.
Nilsen, RSC Advances, 2014, 4, 11876-11883.
- P.-A. Hansen, H. Fjellvåg, T. G. Finstad and O.
Nilsen, Chem. Vap. Deposition, 2014.
UM16
Ferroelastic
hardening in ferroelectric (1-x)Bi0.5K0.5TiO3–xBiFeO3
(x = 0.8, 0.9)
Espen
Tjønneland Wefring*, Kyle Webber¤,
Mari-Ann Einarsrud*, Tor Grande*
* Department of
Materials Science and Engineering,Norwegian University of Science and
Technology,Norway
¤ Nichtmetallisch-Anorganische Werkstoffe, Technische
Universität Darmstadt, Germany
Contact author: tor.grande@ntnu.no (Tor Grande)
Lead-free piezoelectric materials have gained significant attention in
recent years[1]. One of the highly interesting materials is BiFeO3
(BFO) with a high strain and polarization, and possible high
temperature applications due to the high TC. The application of BFO is
though challenging because of a high coercive electric field and a
problematic electrical conductivity [2]. Due to the synthesis
challenges of pure BFO, (1-x)Bi0.5K0.5TiO3
–xBiFeO3
((1-x)BKT - xBFO, x = 0.8, 0.9)has been studied as a model system to
better understand the point defect chemistry and electrical
conductivity of BFO[3].
The high coercive electric field of BFO has been related to point
defects that arise due to evaporation of Bi2O3during
fabrication [4]. Point defects due to this loss may act as pinning
centers for domain walls and hamper wall movement, reducing the
polarization and strain observed for polycrystalline BFO. The hard
ferroelectric properties of BFO have motivated the present study of
ferroelastic properties, possibly related to the same phenomenology. An
advantage of ferroelasticity is that challenges related to conductivity
and dielectric breakdown at high electric field are avoided.
Here we report on a study of aging/hardening in relation to point
defects in BKT-BFO by stress-strain measurements in samples with
different thermal history and with donor doping. Sintered samples were
grinded to a cylinder shape and annealed above TC to relieve residual
stresses. A uniaxial stress up to 800MPa was applied on the samples at
temperatures up to 400°C. Crystal structure and phase
transitions
were examined by high temperature X-ray diffraction, thermal analysis
and dielectric spectroscopy.
Fig. 1: Temperature dependence of
stress-strain curves for 0.8BFO.
The results show that the thermal history of the materials is of great
importance for the stress-strain curve. A furnace cooled sample
(300°C/h) i.e. shows no ferroelastic behavior while an air
quenched
sample demonstrates opening of the stress-strain curve and remanent
strain. The ferroelastic properties were found to vary significantly
with temperature (Fig. 1). It was also found that the crystal structure
and the ferroelastic properties were sensitive to donor doping which
further implies that the point defect chemistry of the material is of
great importance.
References
- J.
Rodel, et al. J. Am. Ceram. Soc., 92, 1153-1177(2009)
- G. Catalan and J. F. Scott. Adv. Mater., 21, 2463-2485
(2009)
- M. I.Morozov, et al.Appl.Phys.Lett.,101 (25),
2529041-2529044 (2012)
- T. Rojac, et al.J. Appl.Phys. 108(7),074107(2010)
UM17
STABILITY
OF CARBON CONDUCTIVE ADDITIVES AT HIGH OPERATING VOLTAGES IN LI-ION
CATHODES
Ann Mari Svensson, Benedicte Eikeland Nilssen, Marita Sætnan, Ahmet Oguz
Tezel,
Department
of Materials Science and Technology, Norwegian
University of Science and Technology (NTNU)
e-mail: annmari.svensson@ntnu.no
Carbon
conductive additives
are important constituents of the Li-ion battery cathodes, as the
oxidic cathode materials used up to now are suffering from too low
electronic conductivities. The next generation, high capacity cathodes
are expected to operate at higher voltages than the current
state-of-the-art cathodes, which will be challenging for the stability
of the carbon conductive additive. It is already know that the anion
PF6- intercalates in graphite at potentials above 4.2 V, and also that
decomposition of electrolyte and exfoliation of graphite due to
co-intercalation of electrolyte may occur [1,2]. At higher voltages all
know electrolytes will oxidize, which in particular cause degradation
of high surface area conductive additives, like carbon black.
The purpose of this study is to compare different carbon conductive
additives with respect to their electrochemical performance at high
cathodic voltages. In addition to conventional graphite powder KS6 and
the carbon black SuperP Li from TIMCAL, a multilayer graphene powder
was included in the study. Electrodes from these materials were then
cycled galvanostatically in coin cells at various rates, and
investigated in 3-electrode experiments with cyclic voltammetry and
impedance spectroscopy. The electrolytes used were 1M LiPF6
in 3:7 EC:DMC or 1:1 EC:DMC.
Results show that the intercalation potential of PF6-
is
slightly higher for multilayer graphene compared to KS6, and also more
reversible. On the other hand, the graphene has a much higher
irreversible capacity loss in the first cycle, indicating more severe
electrolyte oxidation, although the BET surface areas are similar. All
materials showed stable performance upon galvanostatic cycling at 120
mAh/g. Apart from electrolyte oxidation products, no changes to the
electrodes could be observed by SEM after cycling. In-situ X-ray
spectroscopy was performed to obtain more information on the anion
intercalation processes in the multilayer graphene material as compared
to the graphite material. The results were relatively similar for the
two materials, and show that the intercalation of PF6- is only partly
reversible.
References
- J.A.
Seel and J.R. Dahn, J. Electrochem. Soc. 147(3) (2000) p 892.
- W. Märkle, J.T. Coli, D. Görs, M.E.
Spahr, P.Novak, Electrochimica Acta, 55 (2010) p. 2727
UM18
Investigation
of the electrical conductivity in LiAlO2 thin films deposited by Atomic
Layer Deposition
Yang Hu, Amund
Ruud, Ville Miikkulainen, Truls Norby, Ola Nilsen, Helmer
Fjellvåg
Centre
for Materials Science and Nanotechnology and Department of Chemistry,
University of Oslo
P.O. Box 1126, Blindern, NO-0318 Oslo, Norway
Atomic
Layer
Deposition (ALD) allows deposition of highly conformal films on complex
and 3-dimensional (3D) structures. It has been recognized as a suitable
and promising tool to achieve solid-state and 3D designs to obtain
safer and more robust Li-ion battery systems. Recent successful
deposition of Li-containing thin films by ALD [1-3] opened up new
possibilities to develop solid-state electrolytes as well as other
active materials for Li-ion (micro)batteries. For the moment, the
investigation of potential solid-state electrolytes by ALD is still new
[3] and there are currently very few reports on the crucial
characteristic – Li-ion conductivity in such thin films
[4-6],
especially at room temperature. This is probably due to the low Li-ion
conductivity in solid state and the challenges in conducting electrical
measurement on amorphous thin films.
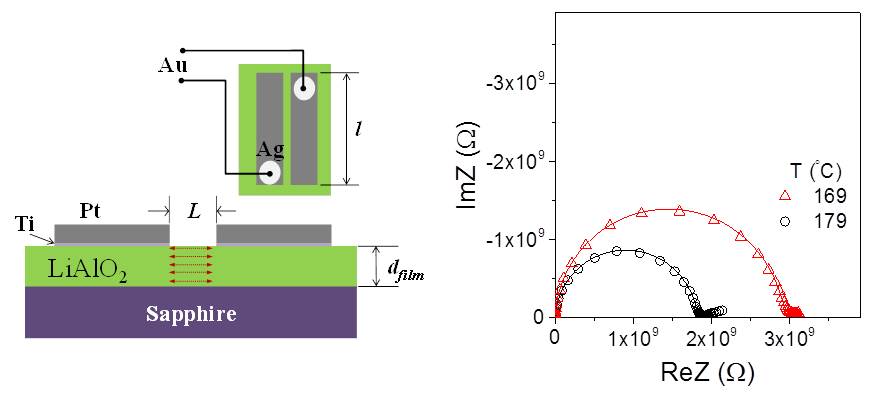
Fig.
1. (a) Geometrical
configuration of in-plane conductivity measurement. (b) Impedance
spectra of 160 nm-thick LiAlO2 thin films on sapphire substrate
obtained by in-plane measurement. The inset shows the equivalent
circuit for data fitting, and the solid lines represent the fitting
results.
LiAlO2 have been deposited by ALD [5]
and were
utilized as barrier and protection layers [5,7] in Li-ion batteries.
This work aims at investigating the conductivity properties of the ALD
LiAlO2 amorphous films in the temperature range
of interest.
Electrical characterizations were carried out by impedance spectroscopy
from ambient to elevated temperature in controlled atmosphere. An
optimized “soft” contacting method is used to make
effective and stable electrode contacts. We applied both in-plane and
cross-plane measurements, depending on the type of substrate. The
conductivities obtained using the cross-plane method are relatively
independent of the film thickness, whilst results from in-plane
measurements exhibit a stronger thickness-dependence. Room-temperature
conductivity in these amorphous LiAlO2 films
could be
readily measured, and the temperature-dependent ionic conductivity
obtained from the two methods are reported and compared. .
References
- M.
Putkonen, T. Aaltonen, M. Alnes, T. Sajavaara, O. Nilsen,H.
Fjellvåg, J. Mater. Chem. 2009, 19, 8767-8771.
- T. Aaltonen, V. Miikkulainen, K. B. Gandrud, A. Pettersen,
O. Nilsen,H. Fjellvåg, ECS Trans. 2011, 41, 331-339.
- O. Nilsen, V. Miikkulainen, K. B. Gandrud, E.
Østreng, A.
Ruud,H. Fjellvåg, physica status solidi (a) 2014, 211,
357-367.
- J. Liu, M. N. Banis, X. Li, A. Lushington, M. Cai, R. Li,
T.-K. Sham, X. Sun, The Journal of Physical Chemistry C 2013.
- T. Aaltonen, O. Nilsen, A. Magrasó,H.
Fjellvåg, Chem. Mater. 2011, 23, 4669-4675.
- J. S. Park, X. Meng, J. W. Elam, S. Hao, C. Wolverton, C.
Kim,J. Cabana, Chem. Mater. 2014, 26, 3128-3134.
- D. J. Comstock,J. W. Elam, The Journal of Physical
Chemistry C 2012, 117, 1677-1683.
UM19
EFFECTS
OF SINTERING ADDITIVES ON BaZr1-XYXO3-δ DENSIFICATION,
STABILITY, AND CONDUCTIVITY
Marie-Laure Fontaine1, Mehdi
Pishahang1, Jonathan Polfus1, Paul
Inge Dahl1, Nahum
Masó-Carcasés2, Anna
Magraso2, Truls Norby2, Rune
Bredesen1
1
SINTEF Materials and Chemistry, Sector for Sustainable Energy
Technology, Forskningsveien 1, NO-0314 Oslo, Norway
2 Department of Chemistry, University of Oslo, FERMiO,
Gaustadalléen 21, NO-0349 Oslo, Norway.
This work reports on the
investigation of BaZr0.9Y0.1O3-
δ (BZY10) and BaZr0.9Y0.15O3-δ
(BZY15) materials for application in Proton Conducting Fuel Cells fed
with biogas. Materials were synthesized by Pechini route or solid state
reactive sintering with Ni, Mg, Mn, Cu, or Zn as sintering additive.
Sintering mechanisms for both routes were studied by in situ optical
dilatometry analysis in controlled atmosphere, thermogravimetric and
differential thermal analysis, high temperature scanning electron
microscopy, in situ X-ray diffraction, transmission electron microscopy
and DFT calculations. This study enabled to identify the effects of
additive types and content as well as temperature on the densification
rate and structure of BZY materials.
Dense
samples were
successfully produced when using Ni and Zn oxides, after sintering in
air at 1500 °C and 1375 °C, respectively. BZY samples
produced
with Cu, Mg, and Mn oxides remain porous up to 1600 °C.
Stability
of Cu-, Ni-, and Zn-containing BZY materials in biogas conditions was
investigated by thermogravimetric analysis in mixtures of 60% H2, 10%
H2O, 10% CO2, 20% Ar, and 800 ppm H2S. While Ni-BZY samples remain
stable under these conditions, both Zn and Cu-doped BZY samples
experienced significant changes, attributed to evaporation and
sulphidation, respectively, of the dopants.
The conductivity of BZY15 was studied by impedance spectroscopy as
function of atmosphere, processing routes and additive contents.
Samples prepared from both routes exhibited similar bulk conductivity
in wet air with an activation energy of ~0.48 eV (Fig 1). In a wet
mixture of 5% H2:95 % Ar, the conductivity of the samples prepared by
solid state increased an order of magnitude compared to that in air,
whereas no change in conductivity was observed in samples prepared by
the Pechini route. These results will be discussed in this presentation.
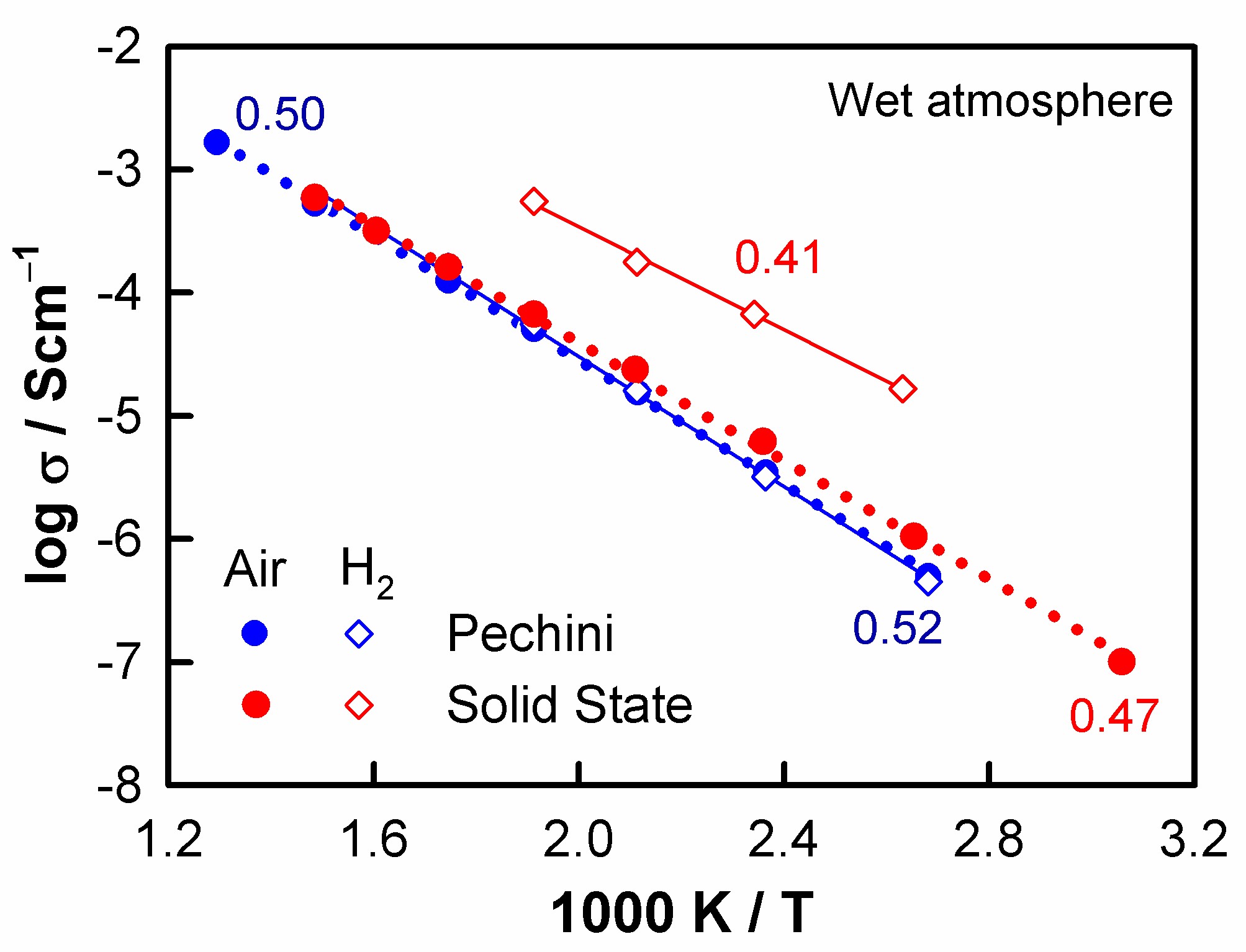
Fig
1. Bulk conductivity Arrhenius plots for BZY15. Numbers represent
activation energies.
UM20
ADDITIVES
IN MAGNESIUM BOROHYDRIDE: LOCAL STRUCTURE AND EFFECT ON REVERSIBILITY
Olena
Zavorotynska, Ivan Saldan,1 Satoshi Hino, Terry Humphries, Stefano
Deledda and Bjørn C. Hauback
Physics
Department, Institute for Energy Technology, P.O. Box 40, NO-2027,
Kjeller, Norway
Magnesium
borohydride is a
particularly interesting material for hydrogen storage due to its light
weight and 14.9 wt% of hydrogen. Up to about 4 wt% have been found to
desorb reversibly below 300oC and at a moderate pressure [1]. For
rehydrogenation of the completely dehydrogenated material, however,
much harsher conditions are needed [2]. In order to improve hydrogen
sorption performance of Mg(BH4)2, a wide range of approaches are
explored, including high energy reactive ball-milling, preparation of
composite materials, dispersion in porous matrix, and addition of
catalysts [3].
|
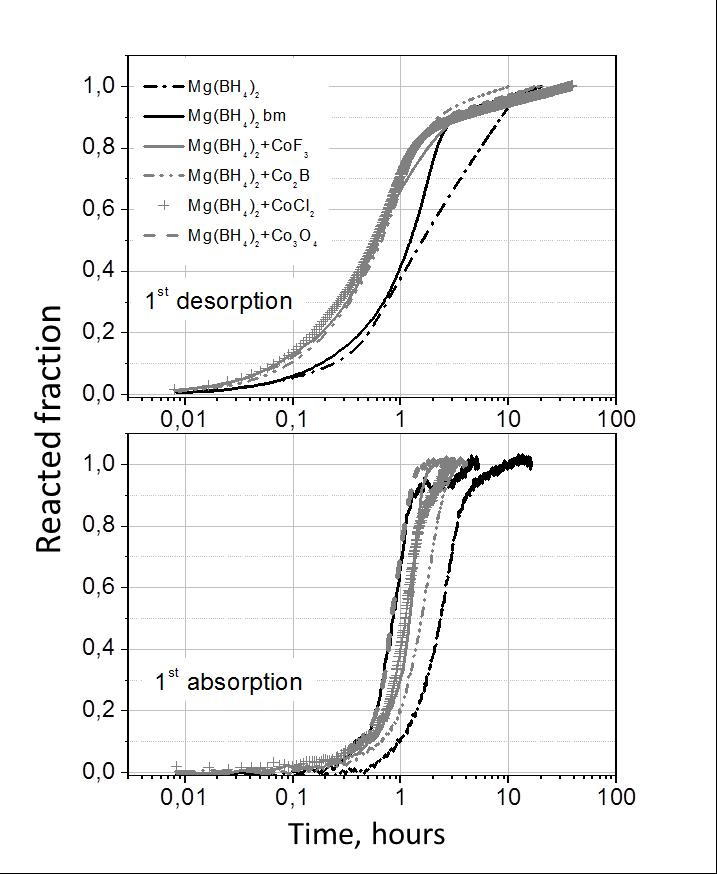
|
Fig. 1. Desorption and absorption isotherms for Mg(BH4)2 as such,
ball-milled (bm), and ball-milled with additives |
Transition metal compounds have been widely explored for enhancing
hydrogen storage properties of complex hydrides, including metal
borohydrides [2, 3]. A number of the additives were shown to reduce
significantly hydrogen release temperature during the first
decomposition of Mg(BH4)2. However, the effect of additives on hydrogen
absorption and further cycling was scarcely studied. In this
contribution we present our recent studies on the effect of transition
metal–based additives in Mg(BH4)2.
A range of the nickel and cobalt compounds was ball-milled with Mg(BH4)2,
and the sorption properties of the composites were studied upon one or
three H-sorption cycles. The desorption and absorption were carried out
at 285oC where Mg(BH4)2
was shown to be
reversible at least for one cycle. The additives were characterized by
x-ray absorption spectroscopy (XAS), which gives an insight into the
local state of metal atoms and the additives composition upon
cycling.
This
work was financed by
the European Fuel Cells and Hydrogen Joint Undertaking
(http://www.fch-ju.eu) under collaborative project
“BOR4STORE” (Grant agreement no.: N°
303428).
References
- G.L.
Soloveichik, M. Andrus, Y. Gao, J.C. Zhao, S. Kniajanski, Int. J.
Hydrog. Energy, 34 (2009) 2144-2152.
- E. Rönnebro, 15 (2011) 44-51.
- H.-W. Li, Y. Yan, S.-i. Orimo, A. Züttel, C.M.
Jensen, Energies, 4 (2011) 185-214.
UM21
POWDER
SYNTESIS AND PROCESSING FROM NANO TO MILLIMETER SCALE
Paul Inge Dahl
& Tommy Mokkelbost
SINTEF
Materials and Chemistry, Dept. New Energy Solutions,
NO-7465 Trondheim
Powders,
ranging from nano-
to millimetre scale, are used for a wide range of applications in the
chemical, metallurgical, pharmaceutical and food industries. SINTEF
Materials and Chemistry performs work related to development of several
technologies where synthesis of powders is essential, e.g. for
catalysts, sorbents, batteries, membranes, fuel cells and medical
applications. Tailoring of both chemical compositions and particle
morphologies through reproducible and scalable processes is essential
for future technology development.
Nanostructured
materials are
gaining widespread use and require new approaches to synthesis, in
particular with respect to increased production while maintaining
proper HSE procedures. Flame spray pyrolysis is an excellent tool for
pioneering development of complex nanomaterials for various
applications, and is also a scalable process already implemented by
commercial powder producers. Use of newly invested flame spray
pyrolysis unit for synthesis of small scale reproducible batches (1-10
grams) of finely dispersed nanoparticles of various inorganic materials
(e.g. mixed oxides) will be presented, along with their potential
application areas. Examples of how varying the precursor solution
system, liquid- and gas flow rates, as well as gas composition
(oxidizing-inert atmosphere) affect the morphology, crystallinity and
chemical composition of the product will be given.
For
lab scale materials to
be attractive for commercialization, viable routes for up-scaled
fabrication and shaping at an industrial level need to be developed,
while maintaining composition and properties. Spray drying is common
route where spherical, dense particles are formed by drying of a
droplets formed by atomizing. The particle size typically scale with
the size of spray dryer due to increased drying time. Other methods
where the final particle size can be easily tailored are granulation,
agglomeration and spray coating. The latter methods can scaled up more
easily from lab to full scale. All these methods result in different
final material properties suitable for a variety of applications.
Results from different projects related to processing of powders using
the above mentioned techniques will be presented – two
examples
given in Fig. 1.

Fig.
1: Manganese oxide micrographs
(TEM/SEM) after
a) flame spray pyrolysis, and b) after
spray
granulation.
HI -
Kjemiens historie
HI1
Norsk
krystallografi gjennom hundre år.
Carl Henrik
Gørbitz
Kjemisk institutt, Universitetet i Oslo

2014
er av FN erklært
som ”The International Year of Crystallography”
(IYCr2014)
som en markering av 100-års jubileet for
røntgendiffraksjon representert ved nobelprisen i fysikk til
Max
von Laue i 1914 etterfulgt av William Henry Bragg (faren) og William
Lawrence Bragg (sønnen) året etter. Laue
påviste
(eller snarere hans assistenter Walter Friedrich og Paul Knipping etter
anvisning fra Laue) diffraksjon fra krystaller i 1912. Senere har det
fulgt en lange rekke viktige oppdagelser. Hva var de, og hvordan ble de
mottatt i sin samtid? I dette foredraget blir det gitt en kronologisk
gjennomgang av banebrytende arbeider på den internasjonale
arena
fra de første strukturbestemmelsene utført av W.
L. Bragg
i 1913 (bl.a. ZnS, diamant og NaCl) til de nyeste studiene av
proteinkomplekser basert på røntgen fri-elektron
lasere
(XFEL). I perioden har strukturkjemien hatt en sterk posisjon i Norge,
der flere markante personer har satt sitt preg på forskningen
ved
våre universiteter. Disse vil bli viet spesiell omtale, med
fokus
på de problemstillingene som har vært sentrale
innenfor
ulike tidsepoker.
HI2
Rekonstruksjon
av destillasjon på 1500-tallet
Fredrik M. Kirkemo
forhenv. Kjemisk institutt, NTNU
I 1557 publiserte den daværende stadsfysikusen i
Frankfurt,
Adam Lonicer (1528 - 1586), en urtebok som ble svært
populær i sin samtid [1]. Urteboken er rikt illustrert med
vakre
tresnitt og beskriver både planter og dyr, men også
destillasjonsteknologien som ble anvendt for å fremstille
medisiner. Boken kan derfor betraktes både som et
oppslagsverk om
planter og dyr, som en farmakologi og som en håndverksbok om
destillasjon. De flotte illustrasjonene og den øyensynlig
store
detalj-rikdommen dannet inspirasjonen til en fullskala rekonstruksjon
av destillasjonsprosesser fra 1500-tallet. Dette prosjektet har
vært gjennomført som en masteroppgave ved
Institutt for
kjemi ved NTNU, som en del av det tverrfaglige Mubil - prosjektet
(museum og bibliotek - et digitalt laboratorie) [2].
Med hjelp fra spesialiserte håndverkere fra
universitetets
verksteder ble det rekonstruert en ovn, utstyr samt det glassutstyret
som behøvdes for å reprodusere renessansens
destillasjonsteknologi.
Målet med rekonstruksjonen var å
undersøke
utstyret og teknologiens effektivitet, målt med moderne
metoder,
samt å utforske synergieffekten av å arbeide med
teksten
parallelt med den faktiske rekonstruksjonen. Rekonstruksjon av
historisk utstyr og eksperimenter i alkymi og kjemi har, som
metodologi, vist seg nyttige i flere nyere studier [3], men har i
større grad vært anvendt i studier av metallurgisk
alkymi
enn iatrokjemisk alkymi [4].
Analysene av det rekonstruerte utstyret viser at noen
hypoteser om utviklingen av destillasjonsteknologi bør
revideres.
Referanser
- Urteboken finnes tilgjengelig digitalt på
http://www.ntnu.no/ub/spesialsamlingene/ebok/kreuterbuch.html
- Mer informasjon om Mubil finnes på
http://www.ntnu.no/ub/omubit/bibliotekene/gunnerus/mubil
- Essays i Holmes, F. L. og Levere, T. H. 2002.
Instruments
and Experimentation in the History of Chemistry. The MIT Press, London.
- Martinón-Torres, M. 2011. Some recent
developments in the historiography of alchemy. Ambix 58 (3). s. 215-37.
HI3
Organisk kjemi i 1830
Ragnar
Bye, Berit Smestad Paulsen og Bjørn Pedersen,
Farmasøytisk og Kjemisk institutt
UiO
Jørgen
Gløersens notater fra
professor Jac Keysers forelesninger i den organiske kjemi i 1830 har
blitt oppbevart på Nasjonalbiblioteket lenge, men vi ble klar
over dem først da kartotekkortene i
Håndskriftsamlingen
ble digitalisert og lagt ut på nettet. Notatene er skrevet
med
gotisk håndskrift som vi har fått transkribert av
Marianne
Kern. Marianne er ikke kjemiker så vi har
gjennomgått og
gjort endringer i teksten der Marianne tydeligvis har
misforstått
enkelte kjemiord.
Gløersen
var 24 år da han skrev disse notatene. Han tok Eksamen artium
i
1826, anneneksamen (Ex. Phil.) i desember 1827 og medisinsk
embetseksamen i mai 1833. Til anneneksamen hadde han bl. a. blitt
eksaminert av Keyser i naturlære dvs. fysikk og kjemi. Det
betyr
at disse forelesningene Keyser holdt, må være et
avansert
kurs i organisk kjemi for medisinerstudenter og
apotekerlærlinger
som bygget på det de hadde lært tidligere.
Berzelius
hadde publisert sin bok i organisk kjemi tre år
før
[1]. Han, og andre kjemikere den gang, trodde at organiske
stoffer bare kunne syntetiseres av en levende organisme og at slike
stoffer ble syntetiseres i spesielle organer. Derav navnet organisk
kjemi. Notatene viser at Berzelii bok har vært kjent av
Keyser,
men bemerkelsesverdig er at de kjemiske formler gitt av Berzelius ikke
er gjengitt i notatene.
Den
organiske kjemi som presenteres i Gløersens
forelesningsnotater
er kjemi knyttet til planter og hvordan man kan fremstille stoffer fra
planter. Mye av det som er skrevet er vanskelig å
forstå
for oss i dag. Vi har derfor lagt til mange, forhåpentligvis
oppklarende kommentarer til både kjemien og botanikken slik
at
dagens lesere kan forstå hva Keyser foreleste for
Gløersen
den gang i 1830.
Planen
var at dette arbeidet skulle vært ferdig i tide til dette
landsmøte, men det rakk vi ikke. Vi håper at et
hefte om
Gløersens notater med våre kommentarer kan
foreligge til
jul i år.
Referanse
- J.J. Berzelius: Lärbok i organiska kemien. (1827)
Eget forlag.
HI4
Tidsskriftet Kjemis historie fra 1904 til 1959
Bjørn
Pedersen
Skolelaboratoriet, Kjemisk institutt UiO
2014-årgangen av tidsskriftet Kjemi kalles
årgang 74,
men egentlig burde det ha vært årgang 111. Det
skyldes at
dagens nummeringen starter i 1941 som første
årgang kalt
Tidsskrift for kjemi, bergvesen og metallurgi. Det avløste
Tidsskrift for kjemi og bergvesen som kom i 20 årganger fra
1921.
Det avløst på sin side Tidsskrift for kemi som kom
i 17
årganger fra 1904. Så vi kan si at tidsskriftet har
kommet
i tre serier: 1. serie fra 1904 til 1920, 2. serie fra 1921 til 1940 og
3. serie fra 1941 til i dag.
|

|
Skolelaboratoriets
samling av tidsskriftene fra 1904 til 2014.
Serie 1 nederst til venstre og serie 2 nederst til høyre.
Resten er
tidsskriftene i serie 3 (foto: Truls Grønneberg).
|
Jeg vil begrense meg i foredraget til første
halvpart av
perioden dvs fra 1904 til 1959 – 55 årganger. I
1959 var
jeg ferdig med studiene og meldte meg inn i Norsk Kjemisk Selskap.
Siden har jeg fått tidsskriftet og lest hvert nummer.
Første nummer i 1904 het ikke Tidsskrift for kemi,
men
Pharmacia, Tidsskrift for kemi og farmaci. Det var utgitt og redigert
av Eivind Koren (1869-1920). Han var utdannet apoteker, men
konsentrerte sitt liv om tidsskrifter og foreninger for kjemi og
farmasi. I 1915 ble tidsskriftet organ for Norsk Kemisk Selskap, gruppe
i Polyteknisk forening som samme år hadde skiftet navn fra
Kjemigruppen i polyteknisk Forening (P.F.).
Jeg vil i foredraget plukke ut høydepunkter fra
tidsskriftet og beskrive de lange linjer i tidsskriftets historie.
KM -
Kvantekjemi og
modellering
KM1
Orbital transformations for the localization of
non-orthogonal
molecular orbitals
Ida-Marie Høyvika,
Poul Jørgensenb and Jeppe Olsenb
aDept. of Chemistry, Norwegian
University of Science and
Technology, Norway
bDept. of Chemistry, Aarhus University, Denmark
Spatially localized Hartree-Fock molecular orbitals have become an
important tool
in the quest
for low scaling post Hartree-Fock methods. The local orbitals enable us
to exploit
the local nature
of electron correlation to reduce computational complexity, which is
essential for
being able to
do calculations on large molecular systems. When dealing with
orthogonal Hartree-Fock
orbitals,
the orbital locality is limited by nodal structure required to fulfill
the orthogonality
constraints.
Hence, there may be a gain in locality by allowing the molecular
orbitals to be non-orthogonal,
i.e., by relaxing our constraint on the molecular orbital overlap
matrix (the metric). We consider
three orbital transformations for localizing non-orthogonal molecular
orbitals; a metric-conserving
transformation, a metric-breaking transformation, and a transformation
which conserves
the trace of
the metric. All of the above transformations conserve the Hartree-Fock
optimization
condition, and
are used to generate spatially more compact Hartree-Fock orbitals for
the use in post
Hartree-Fock
calculations.
KM2
Redox
processes of different transition metal triphenyl corroles and their
impact on corrole aromaticity.
Hugo Vazquez
Lima
University
of Tromsø
The
triphenylcorrole (TPC) is
the simplest triarylcorrole and several metal-corrole complexes have
been characterized. We used previously reported redox potential values
from 9 TPC compounds (MX[TPC] where M = Mn, Fe, Co, Cu, Ag, Pt or Au
and X = C6H5, C6H4CN,
C6H5CN, Cl or P(C6H5)3)
to test the ability of 5 DFT functionals to reproduce their first
oxidation and first reduction potentials. It was found that BP86 and
B3LYP have the best estimation of the experimental data and that most
of these redox processes take place at TPC, only 2 out of 16 happen at
the metal center. Additionally, it was noticed that TPC suffers
geometrical rearrangements to avoid metal-centered redox processes
while oxidized from aromatic [TPC]3- to
antiaromatic [TPC]-.
The geometrical distortions were further analyzed and identified in a
set of 110 crystallized corroles. Trends in corrole geometrical
patterns and its correlation with aromaticity were established.
KM3
Local electric fields and ionization potentials in dielectric
liquids.
Nazanin Davari1, Christopher D. Daub1,
Per-Olof Åstrand1, and Mikael Unge2
1 Department of Chemistry, Norwegian University of Science
and Technology (NTNU), NO-7491 Trondheim, Norway
2 ABB Corporate Research, SE-72178 Västerås,
Sweden
Insulating liquids are often used as a dielectric barrier between two
electrodes in high-voltage equipments and may suffer a breakdown in
high electric fields. Breakdown happens when a conductive plasma
channel, a streamer, is created in the high field regions which
propagates
through the barrier and bridges the gap between two electrodes. This
phenomenon is influenced by the electric field-dependent molecular
properties of the insulating liquid. The ionization potential decreases
with increasing field while the excitation energies remain almost
constant in
comparison to the ionization potential [1-3]. At an excitation energy,
the response of the local field to the external electric field
increases abruptly,
and local field factors have been calculated for liquid benzene. These
changes could affect the number of free electrons at the location of
high
electric fields so that the propagation of streamers can be altered in
dielectric liquids.
References
- N.
Davari, P.-O. Åstrand, S. Ingebrigtsen, and M. Unge,
“Excitation energies
and ionization potentials at high electric fields for molecules
relevant for electrically insulating liquids,” J. Appl.
Phys.,
vol. 113, 143707, 2013.
- N. Davari, P.-O. Åstrand, and T. Van Voorhis,
“Field-dependent ionisation potential by constrained density
functional theory,” Mol. Phys., vol. 111, pp.1456-1461, 2013.
- N. Davari, P.-O. Åstrand, M. Unge, L. E.
Lundgaard, and
D. Linhjell, “Fielddependent ionization potentials and
excitation
energies: implic
KM4
Molecules in strong magnetic fields: non-uniform fields
Erik Tellgrena
aDept. of Chemistry, Centre for
Theoretical and
Computational Chemistry, University of Oslo, Norway
I will discuss recent work on ab initio (primarily Hartree-Fock level)
methods for molecules in finite magnetic fields, with focus on
non-uniform magnetic fields. Non-uniform magnetic fields give rise to
novel static
response properties,
e.g. molecular anapole moments and anapole susceptibilities. Numerical
results
illustrate a connection between anapole susceptibilities and chirality
as well as
the dramatic improvement in basis set convergence offered by London
atomic
orbitals [Tellgren & Fliegl, JCP 139:164118, 2013]. Because
spin
symmetry is
lost, this is also a case where a full treatment requires 2-component
methods
despite the lack of relativistic effects.
KM5
Coupled-cluster theory for molecules in strong magnetic
fields
Stella Stopkowicz, T.Helgaker
aDept. of Chemistry, Centre for
Theoretical and
Computational Chemistry, University of Oslo, Norway
In astrochemistry, very strong magnetic fields play an important role.
Fields of about 100 T to 100 kT arise around white dwarfs while in
neutron stars the fields are even higher (1-100 MT). The interpretation
of observed spectra is hampered by the fact that in laboratory
measurements magnetic
fields can only be produced to up to about 50 T for static fields and
to 1000 T in destructive pulse experiments.
Therefore, theoretical investigations may help in understanding the
substantial influence of strong magnetic fields on the energies and
properties of molecules. Usually, magnetic field-dependence in
quantum-chemical calculations is treated via a perturbative approach
using a Taylor
expansion around the field B = 0. Employing a more rigorous treatment,
however, enables the study of molecules in finite fields of arbitrary
strength and led to findings showing distinctly modified chemical
properties in very strong fields such as a paramagnetic-to-diamagnetic
transition for
closed-shell molecules [1] and a new paramagnetic bonding mechanism in
diatomics [2].
In this work, coupled-cluster capabilities are introduced to the LONDON
program which treats the magnetic field in a non-perturbative manner
and uses London orbitals (GIAOs) in order to ensure gauge-origin
invariance. As Full-CI results indicate an increasing single-reference
character for
higher field-strengths, the performance of truncated coupled-cluster
approaches is expected to be advantageous. We will present results for
energies and magnetizabilities, thereby investigating the influence of
field-strengths, electron-correlation, and the use of GIAOs.
References
- E.
I. Tellgren, T. Helgaker, and A. Soncini, Phys. Chem. Chem. Phys., 11,
5489 (2009).
- K. K. Lange, E. I. Tellgren, M. R. Ho�mann, and T.
Helgaker, Science, 337, 327 (2012).
KM6
Modeling of the adiabatic connection under the influence of
external magnetic fields
Sarah Reimann, Ulf Ekström, Alex Borgoo, Trygve
Helgaker
aDept. of Chemistry, Centre for
Theoretical and
Computational Chemistry, University of Oslo, Norway
When studying electronic systems, the density is a central quantity. It
is of particular interest for magnetic properties, where the
diamagnetic part is solely determined by the ground state density at
zero magnetic field, and only the paramagnetic part explicitly depends
on the external magnetic field. In other words, as long as a method
gives a considerably wrong density, there is little point in fixing the
more elaborate electron correlation entering the paramagnetic part of
the properties, since that one will be prevailed by the error in the
diamagnetic part due to the wrong density.
We therefore study the electron density obtained with several density
functionals, and compare the error made with respect to the density
obtained with Coupled Cluster, in particular CCSD(T). Special emphasis
is put on the comparison with Hartree-Fock.
We show that, in general, the error of the total density
integrated over whole space is smaller for the DFT calculations, which
is reasonable, since integrals like energies and properties is what the
DFT functionals are optimized for. However, pointwise, there exist
regions where DFT performs considerably worse than Hartree-Fock, in
particular near the nuclei. Since the total density always
integrates to the total number of electrons, a huge error in small
regions like the intermolecular axis must result in a corresponding
error in outer regions, which in the end also effects the calculation
of properties.
This is an important starting point for further work on the
optimization of DFT functionals. Apart from the density, we also study
the NMR shielding, and analyze diamagnetic and paramagnetic part
separately.
KM7
High order geometric derivatives in quantum chemistry
calculations:
challenge, solution and implementation
Bin Gaoa
aCenter for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Tromsø-The
Arctic
University of Norway
The evaluation of high order geometric derivatives presents great
challenge in practical
calculations. Both the use of processors and memory need to be
considered carefully
for an efficient evaluation of the large amount of high order geometric
derivatives, in
particular in the case of large molecules. In the current contribution,
I will address this
problem by generating and addressing any non-redundant and non-zero
geometric derivative
on the fly. The proposed scheme here allows one to calculate all
geometric derivatives
effectively by using a large number of processors. Moreover, a
two-level parallelization
(distribution of geometric derivatives and calculations of individual
derivatives/integrals)
and an implementation of distributed-memory matrix (for instance
through ScaLAPACK)
will make it possible to calculate high order geometric derivatives of
large molecules.
KM8
Why is Dithizonatophenylmercury(II) photochromic?
An electronic structure study of the mechanism
Jeanet Conradie,1 Karel G. von
Eschwege,1 Heike Fliegl,2
Espen Tangen,3 and Clemens Woywod4
1 Department of Chemistry, PO Box 339, University of the Free
State, Bloemfontein, 9300, South Africa
2 Centre for Theoretical and Computational Chemistry, Department of
Chemistry, University of Oslo, N-0315 Oslo, Norway
3 High Performance Computing Group, University of Tromsø -
The Arctic University of Norway, N-9037 Tromsø, Norway
4 Centre for Theoretical and Computational Chemistry, Chemistry
Department, University of Tromsø - The Arctic University of
Norway, N-9037 Tromsø, Norway
The
color of dithizonatophenylmercury(II) (DPM) in non-polar solvents, e.g.
in hexane, changes reversibly from orange to blue under illumination
with blue light [1–3]. This photochromic phenomenon
corresponds
to a photoisomerization around a ground-state C==N double bond. We have
performed calculations employing the DFT, TDDFT, CC2 and CASSCF
electronic structure methods in order to answer the following
questions: (i) Which electronic states are involved in the
photoreaction? (ii) Which nuclear degrees of freedom, in addition to
the reactive C==N bond, are activated in this process? (iii) Can we
identify one or more conical intersections that induce internal
conversion processes? (iv) Do spin-orbit coupling effects triggered by
mercury play a role in the rearrangement? In this talk, preliminary
results will be presented. In particular, we show that the reaction
is initialized by excitation of S2 and not of S1.
References
- K. G. von Eschwege, J. Conradie and J. C. Swarts, J. Phys.
Chem. A 112, 2211 (2008).
- H. Schwoerer, K. G. von Eschwege, G. Bosman, P. Krok and J.
Conradie, Chem. Phys. Chem. 12, 2653 (2011).
- K. G. von Eschwege, G. Bosman, J. Conradie and H.
Schwoerer, J. Phys. Chem. A 118, 844 (2014).
KM9
Theory-Assisted Discovery and Development of Z-Selective
Olefin
Metathesis Catalysts
Giovanni Occhipinta,
V. Koudriavtseva, Karl W. Törnroosa,
Vidar
R. Jensena
aDept. of Chemistry, University of
Bergen,
Allégaten 41, 5007 Bergen, Norway
Giovanni.Occhipinti@kj.uib.no
Traditional catalyst discovery is driven by chance and serendipity and
improving the design
of a catalyst usually involves costly and time-consuming experimental
trial-and-error. However,
little by little computational chemistry is becoming more of a guide to
experiment, and this trend is
transforming the way in which catalysts are discovered and developed
[1]. Here we present one
example in which theory has been determinant for the discovery and
improvement of highly Zselective
olefin metathesis catalysts. Achieving such catalysts has been a major
goal in olefin
metathesis for years [2], with reasonably selective catalysts being
obtained only recently [3]. With
the help of density functional theory calculations, we discovered that
non-selective, commercially
available ruthenium-based catalysts easily can be modified to become
highly Z-selective by
replacing one of the two anionic ligands (most often a chloride) with a
sterically demanding
thiolate; see the left-hand side of Figure 1 [4]. We have developed a
comprehensive computational
approach to improve the design of these catalysts. This approach is
based on detailed insight into
the metathesis mechanism and also accounts for catalytic activity and
stability. Using this approach,
we have reached a catalyst (2) with an unprecedented robustness,
tolerating air and acids, from 1 by
replacing the chloride ligand by isocyanate [5]. Further improvements
include increasing the steric
bulk of the aryl group (Ar) in para position of the benzene thiolate,
achieving catalysts giving up to
97 % Z-selectivity in metathesis coupling reactions of common 1-alkenes
(3) [6].
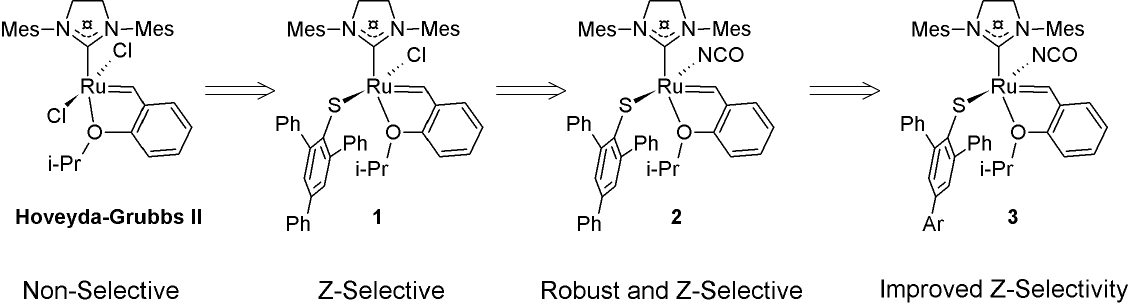
Figure 1: Evolution of Z-selective, thiolate-based ruthenium olefin
metathesis catalysts.
References
- G. Occhipinti, H.-R. Bjørsvik, Jensen, V. R. J.
Am. Chem.
Soc. 2006, 128, 6952. Y. Chu, W. Heyndrickx, G. Occhipinti, V. R.
Jensen, B. K. Alsberg
J. Am. Chem.
Soc. 2012,134,
8885.
- A. Fürstner,
Science 2013,
341, 1357.
- M. M Flook, A. J. Jiang, R. R. Schrock, P.
Müller, A. H.
Hoveyda, J. Am. Chem.
Soc. 2009,
131, 7962. B. K.
Keitz, K. Endo, M. B. Herbert, R. H. Grubbs, J. Am. Chem. Soc.,
2011 ,
133, 9686.
- V. R. Jensen, G. Occhipinti, F. Hansen, Novel Olefin
Metathesis
Catalysts. Int. Patent Appl.
WO 2012032131, 2012. G. Occhipinti, F. R. Hansen, K. W.
Törnroos,
V. R. Jensen J. Am.
Chem. Soc. 2013,
135, 3331.
- G. Occhipinti, V. Koudriavtsev, K. W. Törnroos,
V. R.
Jensen Dalton Trans.,
2014,
43, 11106.
- G. Occhipinti, V. Koudriavtsev, K. W. Törnroos,
V. R.
Jensen to be
submitted. 2014.
KM10
Asymmetric transition metal-catalyzed hydrogenation
ractions:
Insights into the selectivity-determining factors
Kathrin Hopmann
Center for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Tromsø-The
Arctic
University of Norway
This presentation focuses on DFT studies of transition metal-catalyzed
asymmetric hydrogenation reactions. We have investigated several chiral
hydrogenation catalysts to elucidate their mechanistic details and the
factors that govern the experimentally observed enantiomeric excesses
(ee’s) [1-3]. Particular focus will be on alkene
hydroge-nation with iridium complexes exhibiting N,P type ligands
[4]. We show that for this type of complexes, the
selectivity-determining interactions between substrate and catalyst are
governed by strong CH/π type interactions (Fig.1a). Further, we
provide a possible explanation for the experimentally observed
temperature-dependence of the ee.[1,4] The reduced ee observed at lower
temperature is not due to a change in the relative hydrogenation
barriers (∆∆G≠R-S appears unaffected by temperature), but is due
to
an increase in the barrier for isomerization between two isomeric
substrate-catalyst complexes (minor and major, Fig. 1b), which differ
with respect to the alkene-coordination mode. We further present a
small set of benchmark calculations on different iridium-complexes,
showing good performance of the employed computational protocol
(B3LYP-D2/IEFPCM) [1].
a)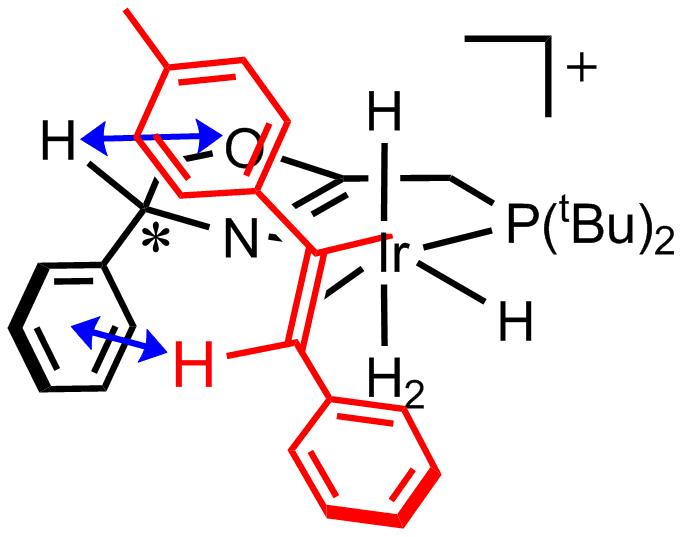 |
b)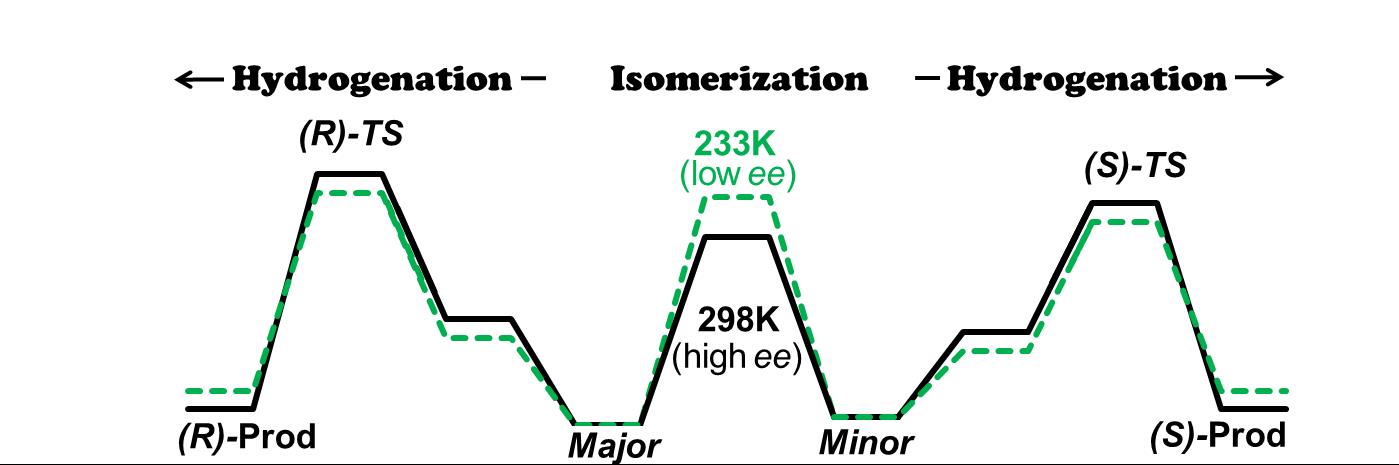 |
Fig. 1: a) Example of selectivity-determining CH/π interactions
(blue arrows) between an alkene substrate (in red) and a chiral
N,P-iridium hydrogenation catalyst (in black). b)Schematic illustration
of computed energy profile. The barrier for isomerization between
alkene-catalyst complexes (minor and major, differing in the alkene
coordination mode) increases at lower temperature, which results in
reduced enantiomeric excess (ee).
References
- Iridium-PHOX-mediated
alkene hydrogenation: Isomerisation influences the stereochemical
outcome, Hopmann, K.H.; Frediani, L.; Bayer, A. Organometallics, 2014,
33, 2790.
- On the Mechanism of Iridium-Catalysed Asymmetric
Hydrogenation
of Imines and Alkenes: A Theoretical Study, Hopmann, K.H.; Bayer, A.
Organometallics 2011,30, 2483.
- Cobalt-Bis(imino)pyridine-Catalyzed Asymmetric
Hydrogenation:
Electronic Structure, Mechanism, and Stereoselectivity. Hopmann, K.H.,
Organometallics 2013, 32, 6388.
- Asymmetric Hydrogenation with Iridium C,N and N,P Ligand
Complexes: Characterization of Dihydride Intermediates with a
Coordinated Alkene Gruber, S., Pfaltz, A. Angew. Chem. Int. Ed. 2014,
53, 1896.
KM11
DFT Adventures on Structure, Reactivity, Development and New
Concepts
David Balcellsa
aCenter for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Oslo, Norway
In this talk I will present a short overview of DFT studies in close
collaboration with experimental groups on the field of organometallic
chemistry and catalysis. These studies focus on several topics
including: 1) characterization of polyhydride iridium clusters
(catalytic hydrogenation);[1] 2) mechanistic studies on catalytic
cross-coupling reactions (synthesis of fine chemicals);[2] 3)
unexpected products in the copper-catalysed oxidation of alkanes
(activation of small inert molecules)[3] and 4) unprecedented
strengthening and deactivation of ancillary ligands in transition metal
complexes.[4]
Figure: Joint theoretical-experimental projects with DFT.
References
- Campos, J.; Sharninghausen, L. S.; Crabtree, R. H.;
Balcells, D. Angew. Chem., Int. Ed., 2014, accepted.
- Hruszkewycz, D.; Balcells, D.; Guard, L.; Hazari, N.;
Tilset, M. J. Am. Chem. Soc., 2014, 136, 7300.
- Conde, A.; Vilella, L.; Balcells, D.;
Díaz-Requejo, M. M.;
Lledós, A.; Pérez, P. J. J. Am. Chem. Soc., 2013,
135,
3887.
- Nova, A.; Balcells, D. Chem. Commun., 2014, 50, 614.
KM12
Nucleophilic substitution reactions of partially hydrated
superoxide anions with alkyl halides
Mauritz J. Ryding, Andrea
Debnárová and Einar Uggerud
Mass Spectrometry Laboratory and Centre of Theoretical and
Computational Chemistry, Department of Chemistry, University of Oslo,
Norway,
The ability of a molecule to donate an electron pair to a substrate and
displace a leaving group during an SN2 nucleophilic substitution
reaction is strongly affected by solvent effects.Superoxide anion, O2-,
is an essential intermediate in the cellular processes of electron
transport, including the respiration chain and photosyntheis, and plays
a key role in the immune defence system of organisms. The
investigatigation of the properties and reactivity of superoxide and
superoxide/water clusters in the gas phase offers insight into the
atmospheric chemistry of the species, and understanding of how O2-
reactivity is moderated by hydration.
This work investigates the gas-phase reactivity of bare O2-
and water
clusters containing it, O2−(H2O)n,
in substitution reactions with
CH3Cl, CH3Br. Besides
studying the degree of hydration on reactivity,
it was of interest to investigate the effect of the leaving group and
to examine the nature of the products. The reactions were
studied
using mass spectrometry.
In order to help assisting the interpretation of the data, we have
conducted quantum chemical calculations of the selected reaction
profiles, including the identification of key intermediates and
transition state structures. In addition, Born Oppenheimer direct
dynamics calculations were conducted to examine the product formation
in detail, with particular emphasis on the possibility of water
molecule transfer during reaction.
KM13
Doing Computational Chemistry faster and bigger
-
opportunities for computational chemistry in the national electronic
infrastructure for science
Espen Tangen a
a Norwegian Supercomputing Centre
(NOTUR), Norway
Abstract:
The Norwegian Metacenter for Computational Science (NOTUR)provides the
national infrastructure for computational science in Norway. Currently,
computational chemistry is one of the major consumers of computer time
on the national facilities.
In addition, we have indications showing that our users in average
publishes in more highly ranked jornals than academic staff that not
are using computational resources.
Thus, the focus of this talk would be split:
I present the national project on direct user community support for
computational chemistry, both in terms of available resources,
possibilities, and the need for better communication!
I also want to present oppurtunities for potential users of our
services. This will include a brief presentation of the different
facilites in Norway, the current list of available software and also
necessary links for applying for access and user training."
KM14
Calculating transport properties from first principles:
thermoelectric materials as a playground
Ole Martin Løvvik a, b
aSINTEF Materials and Chemistry, 0314
Oslo
bUniversity of Oslo, Dept. of Physics, 0316 Oslo
Many devices rely on materials transport properties like electrical
resistivity, heat conductivity, and the Seebeck coefficient. (Given a
temperature difference, the Seebeck coefficient determines the
resulting voltage between two ends of a material.) This is particularly
so for nanoscale energy conversion technologies, which are likely to
change radically how we generate, transport, and use energy in our
daily lives. One example is thermoelectric materials, which can be used
both for heating/cooling purposes through the Peltier effect or
electricity production from waste heat or renewable sources through the
Seebeck effect (Fig. 1).
Prediction of such transport properties is challenging, and it is only
recently that electronic structure
calculations have been available for calculating some of them without
adjustable parameters. We are
in this presentation going to look at what has been achieved so far,
and what remains before reliable
predictions about realistic materials can be expected.
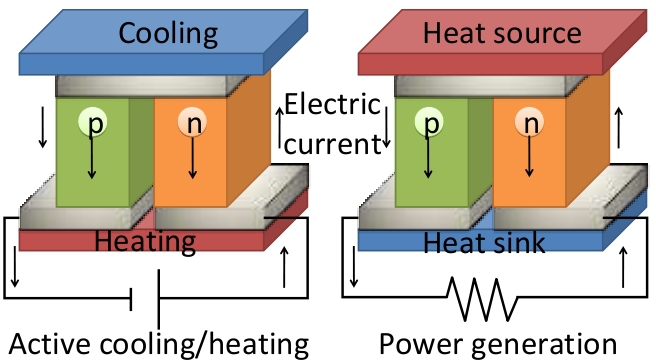
Figure 1: Thermoelectric materials can be used for cooling or heating
through the Peltier effect
(left) or for production of electricity through the Seebeck effect
(right). Their charge carriers are
either electrons (n-doped) or holes (p-doped).
KM15
Chemical structure from photoelectron spectroscopy
Knut Børve a
aDept. of Chemistry, University of
Bergen, Norway
In X-ray photoelectron spectroscopy (XPS) the ionization
energy
of inner-shell electrons is measured by means of soft, monochromic
X-rays. The radiation and hence the relevant ionization energies appear
on an energy scale of 100-1000 eV (104-105 kJ/mol), with an
experimental energy resolution of about 0.1 eV (10 kJ/mol).
Thus,
the spectroscopic process appears on a much coarser energy scale than
most aspects of chemical structure. Nonetheless, by combining
theoretical models of the fine structure in X-ray photoelectron spectra
with state-of-the-art experimental spectra, workers have been able to
extract a wide range of chemical structure information from this, until
recently, unexpected source. The presentation aims to exemplify and
explain different mechanisms whereby photoelectron spectra may provide
information on interatomic distances. In concluding, some remarks will
be offered on the possible future role of XPS as a structure method in
chemistry.
KM16
Computational determination of a mechanism for silicon
island
formation in SAPO materials
Ole
Swanga,b,
Stian Svelleb, Sami Malolab,
and Torstein
Fjermestadb
a aSINTEF Materials and Chemistry, P.
O. Box 124
Blindern, 0314 Oslo, Norway; e-mail: ole.swang@sintef.no
bbCRI inGAP, Dept. of Chemistry, U. of Oslo,P.
O. Box 1030
Blindern, 0315 Oslo, Norway
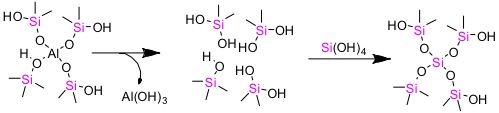
Scheme 1:Al/Si substitution
SAPOs are microporous, crystalline aluminophosphates in which some
phosphorus atoms are substituted by silicon. The substitution
necessitates the presence of extraframework cations; when these are
protons, a solid acid results. SAPOs have important applications in
catalysis. Over time, the silicon atoms move through the material,
forming silica moieties called silicon islands. While a thermodynamic
driving force for this process has been experimentally established,
this is undesirable from a catalytic point of view, and the mechanism
for the reaction has been unknown.
Based on large-scale periodic density functional theory calculations,
we propose a mechanism for silicon island formation. Firstly, silicon
and aluminium atoms are hydrolyzed to Si(OH)4 and Al(OH)3(H2O) by water
molecules in four consecutive steps (Scheme 1, Figure 1). Then, P in
the crystal lattice may be exchanged with Si in hydrolysis reactions
involving water and Si(OH)4. While we cannot rule out other mechanisms
on the basis of these results, the proposed mechanism is
thermodynamically and kinetically feasible.
Figure 1: Si(OH)4 and hydrogarnet defect formed by 4-step hydrolysis.
Acknowledgments:
S. M. and T. F. acknowledge postdoctoral fellowships
from the Research Council of Norway (RCN) under the KOSK 2 program. The
authors thank prof. Karl Petter Lillerud and Dr. Mahsa Zokaie for
valuable discussions, and the NOTUR program of the RCN for a generous
grant of computing resources.
KM17
Defect properties of functional oxides from first principles
Tor Svendsen Bjørheim a
aDept. of Chemistry, University of
Oslo, Norway
KM18
Modulation of protein function by micro-solvation effects:
the
puzzling case of cis-retinal binding in CRALBP
Michele
Cascella,1
Achim Stocker,2 Rachel E. Helbling,2
and Christin S. Bolze2
1 Department of Chemistry, and Centre for Theoretical and
Computational Chemistry
(CTCC), University of Oslo, Norway
2 Department of Chemistry and Biochemistry, University of
Bern, Switzerland
In all biological phenomena involving protein-ligand interactions,
solvation plays crucial effects, both in the thermodynamics and in the
kinetics of binding. In this seminar, I will present how combined
computational and experimental data reveal peculiar and unexpected
properties of binding of cis-retinoids to cellular retinaldehyde
binding protein (CRALBP) and its mutant R234W, associated to Bothnia
retina dystrophy disease. Molecular dynamics simulations show that
residual hydration in the large hydrophobic binding cavity plays a
major role for recognition and binding of the substrates. Due to the
absence of strong directional contacts, multiple conformations of the
ligands inside the cavities of the different proteins are possible at
room temperature for different ligands. The micro-solvation pattern
characteristic of 9-cis-retinal binding in CRALBP evidenced by our
simulations is responsible for a newly discovered secondary thermal
isomerase activity, which may be connected to biological processes
other than imaging forming.
KM19
Reaction path sampling using rare event simulation
techniques
Titus van Erp a
aDept. of Chemistry, Norwegian
University of Science and
Technology, Norway
KM20
Accurate QM/MM made cheaper: a hybrid approach to calculate
polarizable embedding potentials
Maarten Beerepoot a,
Nanna Holmgaard Listb, Arnfinn Hykkerud Steindala,
Jógvan Magnus Haugaard Olsenc,
Kenneth Ruuda
and Jacob Kongstedb
aCenter for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Tromsø-The
Arctic
University of Norway, maarten.beerepoot@uit.no
b Department of Physics, Chemistry and Pharmacy,
University
of Southern Denmark, DK-5230 Odense M, Denmark
c Laboratory of Computational Chemistry and
Biochemistry,
École Polytechnique
Fédérale
de
Lausanne, CH-1015, Lausanne, Switzerland
The balance between accuracy and efficiency is of central importance in
computational chemistry, in
particular in the calculation of accurate molecular properties at a
reasonable cost. Multiscale modeling
deals with this challenge by treating a central part of the molecular
system (e.g. a molecular probe or
the active site of a protein) with an accurate method while describing
the surrounding (e.g. the rest of
the protein or a solvent) with one or more approximate methods. In
polarizable embedding approaches,
the surrounding is modeled by a potential containing charges and
polarizabilities. The most accurate
and expensive way is to derive these parameters from quantum mechanical
(QM) calculations for every
molecule or protein residue separately. The cheapest and most
approximate way is to take only atomic
charges from a pre-parametrized force field such as OPLS. The objective
of our study is to find a
balance between accuracy and efficiency by combining QM-derived
parameters for the innermost
molecules with molecule-specific average parameters for the molecules
further away. We will show
results for optical and vibrational properties of molecules in
different solvents. The approach will be
extended to proteins and lipids in the future to allow for accurate and
efficient modeling of vibrational
and fluorescent probes in biomolecular systems.
KM21
Thermal conductivity of carbon dioxide from non-equilibrium
molecular dynamics: a systematic study of several common force fields
Thuat
T. Trinha,
Thijs J.H. Vlugtb and Signe Kjelstrupa,
b
a 1Department of Chemistry, Norwegian
University of
Science and Technology, Trondheim, Norway
b Department of Process and Energy, Delft
University of
Technology, Delft, Netherlands
Carbon dioxide (CO2) has an important impact on the climate and is
therefore widely studied. Huge efforts are being made, for instance to
reduce emissions of CO2 to the atmosphere, by capture- and
sequestration techniques. In that context, membrane separation
techniques are needed, at high as well as low temperatures.
Fossil-fueled power systems, natural gas processes or production of
hydrogen gas includes all high-temperature separation technologies. The
thermal conductivity of CO2 is needed for process modelling in these
processes.1, 2
We report a systematic investigation of the thermal conductivity of
various three-site models of carbon dioxide (CO2) using nonequilibrium
molecular dynamics in the temperature range 300 -1000K and for
pressures up to 200 MPa. A direct comparison with experimental data is
made. Three popular CO2 force fields (MSM, EPM2 and TraPPE) and two
flexible models (based on EPM2) were investigated. All rigid force
fields accurately predict the equation of state for carbon dioxide for
the given range of variables. They can also reproduce the thermal
conductivity of CO2 at room temperature and predict a decrease of the
thermal conductivity with increasing temperature. At high temperatures,
the rigid models underestimate the thermal conductivity.
References
- Trinh, T.; Bedeaux, D.; Simon, J.-M.; Kjelstrup,S. Chem. Phys. Lett.
2014,
612, 214.
- Trinh, T. T.; Vlugt, T. J.; Hagg, M. B.; Bedeaux, D.;
Kjelstrup,
S. Front Chem
2013,
1, 38.
KM22
Divide wisely and conquer accurately: a strategy to please
chemists who are notorioulsy demanding customers
Luca Frediani a
aCenter for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Tromsø-The
Arctic
University of Norway
KM23
Molecular properties in the random phase approximationy
J. Rekkedal, K.R. Leikanger, M.F. Iozzi, S. Coriani, A. M.
Teale, T. Helgaker, Thomas
Bondo Pedersen
Center for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Oslo, Norway
The random phase approximation (RPA) to the electronic correlation
problem has undergone a revival in the past decade in the quantum
chemistry
and materials science communities [1-3]. Providing a computationally
a�ordable and parameter-free description of dispersion interactions for
metals as well as insulators, RPA is particularly attractive as an
alternative to semi-empirical dispersion-corrected functionals in
density-functional theory. In this talk, a Lagrangian reformulation of
the RPA correlation energy will be presented, paving the way for
calculations of forces and other molecular properties.[4]
References
- A. Hesselmann, A. Görling, Mol. Phys. 109, 2473 (2011).
- H. Eshuis, J.E. Bates, and F. Furche, Theor. Chem. Acc.
131, 1084 (2012).
- X. Ren, P. Rinke, C. Joas, and M. Sche�er, J. Mater. Sci.
47, 7447 (2012).
- J. Rekkedal, S. Coriani, M.F. Iozzi, A.M. Teale, T.
Helgaker, and T.B. Pedersen, J. Chem. Phys. 139, 081101 (2013).
KM24
Cusp based DFT functionals
Espen Sagvolden, Erik I. Tellgren, Ulf E.
Ekström, Trygve U. Helgaker
Center for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Oslo, Norway
Cusp-based correlation functionals model the density-functional-theory
correlation energy by use of a model wavefunction constrained to the
same density as the Kohn-Sham wavefunction, but satisfying the
electron-electron cusp condition. After summing up, expressions can be
rendered as density functionals. The Lee-Yang-Parr (LYP) correlation
functional is a cusp-based correlation functional, based on the
Colle-Salvetti expression. We will discuss the LYP and Colle-Salvetti
expressions and our efforts to rectify their shortcomings.
KM25
Optimisation problems from exact DFT
Ulf Ekström a
aCenter for Theoretical and
Computational Chemistry
(CTCC), Department of Chemistry, University of Oslo, Norway
Lieb's formulation of exact density functional theory
gives rise to optimisation problems that are either constrained or
non-smooth.
These two complications makes the problems much harder than the ones
usually
encountered in quantum chemistry. I will show how non-smoothness is can
be
related by duality to constrained optimisation, and discuss some new
algorithms
developed at the CTCC in Oslo for non-smooth problems.
Finally I will discuss the
geometrical interpretation of the chemical
Aufbau principle, and the connection to the SCF procedure and the DIIS
algorithm of Pulay.
MA -
Matkjemi
MA1
GMO i maten vår – et glimt inn i
fremtiden
Askild
Holck, Seniorforsker
Nofima AS
Hovedpunkter
Genmodifiserte planter (GMP) benyttes til å øke
produktiviteten i landbruket. Dyrkningsarealet for GMP globalt er 175
mill. hektar. De viktigste avlingene er soya (79 % av
verdensproduksjonen), mais (32 %), bomull (70 %) og raps (24 %). Disse
plantene har fått bygget inn ugressmiddeltoleranse og/eller
insektresistens. En rekke nye genmodifiserte planter med andre
egenskaper som tørketoleranse, salttoleranse,
beta-carotenproduksjon og endret oljesammensetning er under utvikling.
Abstract
I år 2050 kommer vi til å være 9
milliarder mennesker på jorden. For å sikre verdens
befolkning nok mat må produksjonen fordobles innen 2050.
Økt produktivitet i landbruket er fortsatt
nødvendig. I planteforedling benytter man en rekke
strategier som gir genetiske endringer i kulturplanter og dermed gir
dem ønskede egenskaper. Genteknologi er én av
mange slike teknikker. Ønsket DNA føres da inn i
planten ved hjelp av bakterier eller ved bombardering med små
metallkuler. Resistens mot plantesykdommer og insektangrep samt
toleranse overfor herbicider er den mest kostnadseffektive
måten å øke produktiviteten
på. De første genmodifiserte plantene (GMP) ble
tilført nettopp slike egenskaper. Fra starten i 1996 har
dyrkningsarealet av GMP økt voldsomt til 175,2 mill. hektar.
De 5 største produsentene av GMP er USA, Brasil, Argentina,
India og Canada. De viktigste avlingene er soya (79 % av
verdensproduksjonen), mais (32 %), bomull (70 %) og raps (24 %). Mange
GMP har flere endrede egenskaper. Andre nye GMP er under utvikling for
eksempel: trehaloseproduserende ris som er salt- og
tørketolerant; tørketolerant mais; golden rice
som produserer beta-caroten og kan avhjelpe A-vitaminmangel i u-land;
insektresistent Brinjal (aubergine) i India;
tørråteresistent potet og dodre som inneholder
fiskeolje. I tillegg utvikles det GMP for produksjon av medisiner
(insulin), phytoremediering og biodrivstoff. GMP har oppført
seg som forventet og dyrkningsarealet for GMP forventes å
øke videre i årene som kommer.
MA2
Risikovurdering av GMO-produkter
Audun
H.
Nerland, Professor
Universitetet i Bergen
Hovedpunkter:
Foredraget vil ta for seg hvordan risikovurderinger av GMO blir
utført i dag, men også sette dette inn i en
større sammenheng, både vitenskapelig, etisk og
politisk. Kan bruk av GMO medføre lavere risiko enn bruk av
tilsvarende konvensjonelle organismer? Vil en for restriktiv
holdning i forhold til GMO være uheldig for Norge i det lange
løp?
Abstrakt
Ifølge den norske genteknologiloven er alle
”mikroorganismer, planter og dyr hvor den genetiske
sammensetning er endret ved bruk av gen- eller
celleteknologi” definert som GMO, uansett hva, eller hvor mye
av det genetiske materialet som er endret. Generalisering når
det gjelder risikovurdering av GMO er derfor ut fra et faglig og
vitenskapelig synspunkt, helt ulogisk. Risikovurderinger,
både angående bruk av GMO som mat og når
det gjelder miljøinteraksjoner, blir derfor
gjennomført for hver enkelt GMO som blir utviklet
(”case by case”). Man ser på hvilken
genetisk modifisering som er gjort, og hva det har som konsekvenser for
den aktuelle organismen sine egenskaper.
I svært mange tilfeller kan utgangsorganismen i seg selv ha
uheldige egenskaper. Ukokte poteter, for eksempel, inneholder
giftstoffer som vil være skadelig hvis man spiser for mye, og
erter kan gi enkelte personer allergiske reaksjoner. Risikovurderinger
blir derfor gjort komparativt. Det vil si at man vurderer om den
aktuelle GMO medfører endret risiko i forhold til
utgangsorganismen (”the conventional counterpart”).
Det samme prinsippet gjelder også miljømessige
risikovurderinger.
Et paradoks når det gjelder risikovurderinger er at
definisjonen av GMO ikke nevner noe om hvorvidt de samme genetiske
forandringene kan oppnås ved bruk av tradisjonelle
avls/foredlings-metoder (som også kan inkludere bruk av
bestråling eller mutagene kjemikalier). Logisk sett burde
alle ”ny-utviklede” organismer bli vurdert ut fra
de genetiske/fenotypiske forandringene som har skjedd, uansett hvilken
metode som er brukt for å oppnå disse
forandringene.
Ved risikovurderinger av GMO burde man også ha sett det hele
i en større sammenheng. Hvilken konsekvens vil det ha hvis
man ikke tillater bruk av en gitt GMO? For eksempel kan
alternativet til bruk av en insekts-resistent GM-plante være
bruk av stor mengder pesticider.
MA3
Tradisjonell avl for bedre geitmelkskvalitet
Knut Erik
Grindaker, Fagsjef
Tine
FoU
En forutsetning for å øke innsatsen på
anvendelse og økt bruk av geitemelk er mild og stabil
kvalitet gjennom året. Tidlig på 2000-tallet var
ikke dette på plass og det ble igangsatt arbeid med
hovedmål å bedre denne situasjonen. Hovedproblemet
var at råstoffet i deler av året var beskt og
harskt. Dette skyldes i stor grad for høyt innhold av frie
fettsyrer i melka, et resultat av fettspalting.
Det ble igangsatt flere prosjekter for å finne mulige tiltak
for å løse problemene på
råstoffsiden. Det ble et gjennombrudd når en fant
sammenhenger mellom kaseingenstatus i geitene og hvor disponert melka
var for fettspalting. Det ble besluttet bevisst å avle
på geiter som var disponert for å produsere alfa
S1-kaseinet. Det ble avlet på norske melkegeiter som hadde
denne egenskapen. I tillegg ble det importert bukkesæd fra
fransk Alpin-geit. Denne geiterasen er kjent for å produsere
alfa S1-kaseinet samt å ha en mild smak.
Det har blitt en formidabel bedring av råstoffkvaliteten,
innholdet av frie fettsyrer er redusert til en tredjedel av
nivået vi hadde i 2005. I tillegg til dette har vi
oppnådd bedre ystingsegenskaper i
melka.
Hovedpunkter
- Bakgrunnen for avlsmessige tiltak
- Avlen hadde hovedfokus på kaseingenstatus i
geitene
- Gjennomføring av avlsprogrammet
- Resultater og status i 2014
MA4
Dyrking av grønnsaker i forskjellig klima-hva
betyr
det for innholdsstoffene?
Gunnar
Bengtsson, Seniorforsker
Nofima AS
De fleste
kulturplanter går ikke å dyrke i alle klimaer,
fordi de ville forgjengerne er tilpasset bestemte voksesteder. Innen
områder der hver art kan dyrkes er det dog forskjeller i
klima som gir forskjeller i kvalitet. Temperaturen er den viktigste
faktoren, deretter kommer lys og vann, men vann er normalt ikke
begrensende for grønnsaker grunnet kunstvanning. Mekanisk
stress fra vind og hagl har også betydning. I nord har
sollyset lavere intensitet av UV-stråling og et
større forhold mellom mørkrødt og
rødt lys, samtidig som lysperioden kan være opptil
24 timer. Alle disse forhold kan påvirke nivået av
innholdsstoffer som bestemmer sensorisk og helserelatert kvalitet:
Polyfenoler, karoteno¬ider, glukosinolater, fettsyrer, sukker,
terpener, vitaminer, etc. Likevel må man alltid ta
utgangspunkt i de nivåer som er genetisk bestemt i ulike
grønnsaksorter. I tillegg kan sortene ha ulik respons for
klimafaktorer. Variasjonen i været fra år til
år kan iblant gi en større effekt på en
grønnsak enn fra ulike klima innen
dyrkingsområdet, f eks. mellom Nord- og Sør-Norge.
Ved tidl. Matforsk ble det forsket på kvalitet av gulrot i en
nord-sørgradient. Lavere temperatur ga søtere og
sprøere, men bleikere gulrøtter. Dette samsvarer
med et lavere innhold av sukrose og karotener og et høyere
innhold av glukose og fruktose. Både før og etter
innhøsting kan mekanisk stress gi bitre gulrøtter
med høyt innhold av isokumariner (6 methoxymellein etc.) og
polyacetylener (falcarindiol etc.). Altfor tett pakning gir
gulrøtter med emmen smak, på grunn av anaerob
metabolisme. På basis av disse resultatene er det utviklet et
produkt som heter Smaksgulrot.
Mange kålvekster klarer bare kjølige betingelser.
I Syd-Europa kan de dyrkes om vinteren, men klarer ikke varmen om
sommeren. Derfor eksporterer faktisk Norge brokkoli og
blomkål til Syd-Europa om sommeren, mens en rekke
kålvekster importeres derfra resten av året. Ved
Nofima har vi i samarbeid med Bioforsk og NMBU nå avsluttet
et prosjekt på effekter av betingelser både pre- og
postharvest for kålvekster («Northern
vegs», støttet av Norges forskningsråd
og Fondet for forskningsavgift på landbruksprodukter). Et
semi-feltforsøk på brokkoli fra Spania til
Nord-Norge er gjennomført i tillegg til flere
forsøk i klimakammer. Resul¬tater fra disse
forsøkene, men også fra postharvest
lagringsforsøk med behandling med UV-stråling og
synlig lys ved ulike temperaturer, vil bli presentert. For ulike
klimaer hadde brokkoli ulik tekstur, utseende og smak. Av
innholdsstoffer i brokkoli ble flavonoler påvirket mest,
deretter ulike glukosinolater på en kompleks måte,
mens vitamin C var lite påvirket.