
Fellesarrangemetet står først, ellers er foredragene nummerert etter faggruppe (alfabetisk):
E - Ernæring og Næringsmiddelteknologi
Km - Kvantekjemi og modellering
M - Makromolekyl- og kolloidkjemi
Dette dokumentet oppdateres etterhvert som abstractene kommer inn.
In
many cases in
vivo diagnostics make the invisible visible. Medical imaging
utilizes most
of the electromagnetic spectrum and includes such diverse technologies
as X-ray/CT, MRI, Ultrasound and Nuclear
medicine (SPECT and PET). The
different imaging techniques have different
characteristics which
determine where in the diagnostic
process they are used. Effective in vivo diagnostics
for X-ray/CT, MRI, Ultrasound and Nuclear medicine have been developed
for diagnosis
of diseases within the cardiovascular,
oncology, neurolgy and pulmonology fields.
Different types of diagnostic information
(anatomical,
functional or molecular) can be obtained depending on the techniques
applied.
Early diagnosis of disease is utmost important. This usually requires
detailed
understanding of the disease process itself. Nuclear
medicine techniques have the required
sensitivity to image at the molecular level which makes early
intervention
possible, may guide therapy and disease follow-up and improve therapy
outcome.
Attributes of high performance in
vivo diagnostics are high diagnostic efficacy, high safety
and
high user friendliness. The chemistry to achieve these challenging
goals must
start with knowledge of
what contrast
parameter to impact as well as understanding the disease target.
Furtermore,
being developed for the commercial market other aspects like synthesis
scale-up, production costs and environmental aspets are very important,
especially regarding contrast media for X-ray/CT which are produced in
ton
scale and where a single injection into a human contains several grams
af the
active ingredient.
The short halflife of the radioisotopes used
in
nuclear medicine has
made it necessary
to develop automated synthesis platforms with plug-in chemistry
cassettes as
well as an automated product quality control system. Effective
syntheseis of 18F
labelled agents has been developed.
Using 3He, 129Xe or 13C in its hyperpolarized state has given the basis for a new type of MRI contrast media. Hyperpolarized 13C labelled small molecules may give metabolic information relevant for diagnosing certain diseases.
A Healthy, Wealthy, Sustainable World deals with the things that people now regard as essential to a modern lifestyle: a secure food supply, clean water, transport fuel, polymers, a comfortable home – and even sports equipment. All these will be put at risk in a world where there are yet more people aspiring to the same high standard of living, but where we no longer rely on fossil fuels. This talk shows that sustainability is possible, but only if we embrace the benefits which chemistry can bring and attract young people to want to study and research our science.
Verneutstyr og betydning av denne har gjort store fremskritt
de siste 30 årene.
Skrekkhistoriene fra 70- og 80-tallet, der industriarbeidere arbeidet
med maling og løsemidler med en enkel fille rundt munnen, og
som fungerte som sniffeklut sitter fremdeles langt inne for oss som
arbeider med dette til daglig. Entreprenører rev bygg med
asbest og eternitt plater uten å vite omfanget av risikoen de
utsatte seg for. Problemet med skade på åndedrett
er at symptomene kan komme årevis etter at skaden
oppstår. Selv om vi har kommet en lang vei, er det fremdeles
utfordringer bransjen, nye teknologier fører også
til nye risikovurderinger. 25 % av all kreft er lungekreft, dette alene
bør være motivasjon nok for økt fokus
på disse områdene.
De siste 10 årene har vi opplevd en eksplosjon i antall
tilbydere av verneutstyr. Etter hvert som antall produkter og
leverandører øker, kan det være
vanskelig å orientere seg om hva som er det riktige for
nettopp ditt bruk. Tenotec er stolte av å være den
første aktøren som importerte og videreformidlet
motorisert og trykkluftsdrevet åndedrettsvern her i landet.
Dette var tilbake i 1976, og man kan trygt si at konkurransen har
økt siden den gang. Vi tilbyr det mest moderne utstyret
på markedet, og vi skreddersyr utstyret som passer den
enkelte bedrifts behov. Våre kunder er alt fra offshore,
entreprenører, sveiseverksteder, skipsverft,
overflatebehandling, landbruk og privatpersoner.
I dag er vi fremdeles Norges største leverandør
av motorisert og trykkluftsdrevet åndedrettsvern, og mye av
arbeidet vårt består fortsatt av å
informere om skadene man kan få og viktigheten av riktig
utstyr for nettopp din risikosituasjon.
Da jorden består av aske fra døde stjerner, har radioaktivitet vært en integrert del av vår natur fra tidenes morgen. Dette skyldes radionuklider med meget lange halveringstider; særlig uran og thorium kjedene som gir opphav til alle elementer tyngre enn bly og vismut, og kalium isotopen 40 K som er en naturlig bestanddel av all kalium. Uranmengden er ca halvert i forhold til da jorden ble skapt. Kosmisk stråling produserer også radioaktive isotoper av karbon (14C) og hydrogen (tritium, 3H).
I tillegg til naturlig forekommende radioaktivt materiale
(NORM), har menneskelig aktivitet bidratt til utslipp av kunstig
produserte radionuklider, spesielt virksomheter knyttet til
”nuclear weapon and fuel cycles”, men
også bruk av radioaktive elementer i ikke-nukleær
virksomhet, som f. eks medisinsk diagnose og terapi.
Konsentrasjon og radionuklidesammensetning i naturlige
prøver (vann, jord/sediment, planter/organismer/dyr og
mennesker) varierer med hensyn på prøvetype,
innsamlingssted, årstid etc. I jordprøver fra
Vestlandet, særlig i Bergensområdet finner vi de
høyeste nivåer fra prøvesprengingene, i
Valdres og Nord-Trøndelag finner vi de høyeste
nivåer av Tsjernobyl nedfallet, i alunskifer og granitt
områder finner vi de høyeste nivåer av
uran, arsen samt en rekke tungmetaller, og i thorium-rike
områder som Fen i Telemark har vi
gammastrålenivåer blant de høyeste i
Europa. Selv om alle prøver vil inneholde radionuklider er
nivåene ofte svært lave. Vi har imidlertid meget
avansert målemetoder basert både på
strålingsegenskaper og nuklidesammensetning som
medfører at vi kan bestemme konsentrasjoner ned i femtogram
området, dvs ”finne nålen i
høystakken”. Likevel må vi
ofte benytte store prøvevolumer, for eksempel trengs 200
– 400 l havvann for å kvantifisere plutonium.
Strålevernloven av 2000 omfatter stålevern for
både mennesker og miljø. I følge de nye
forskriftene til Forurensningsloven (2011) er også
radionuklider inkludert i miljøgiftbegrepet. Begge lovene
innebærer utfordringer både for forskning og
myndighetsutøvelse. Vi har ingen effekt enhet for biota
(Sievert enhet gjelder bare for mennesker) og vi mangler vitenskapelig
grunnlag for å fastsette grenseverdier for miljø
nasjonalt og internasjonalt. Forurensningsloven innebærer at
avfall fra ikke-nukleær industri for eksempel olje - og gass
industri, gruvedrift, treforedling etc. kan, om NORM nivåer
er høyt, klassifiseres som radioaktivt avfall. Tunnelgraving
i alunskifer områder kan innebære at stein ikke
bare kan dumpes, men må behandles som radioaktivt avfall.
Foredraget vil gi en oversikt over radioaktive kilder som
bidrar til forurensning og vil legge vekt på utfordringer
knyttet til håndtering av radioaktive prøver,
analyser, og konsekvensvurderinger.
I foredraget omtales kort syrenes kjemi og de åpenbare analytiske fordelene sett i forhold til de velkjente risikomomentene ved bruk av de to syrene diskuteres. Ikke minst legges det vekt på hva man på forhånd kan gjøre for å minske farene mest mulig. Ulike førstehjelpstiltak – hvis uhell skulle skje – omtales også.
Mikrobølgeovner blir stadig mer utbredt i
laboratoriene. Dette er blitt et nyttig verktøy i
dekomponering av prøver før analyse. Slike
systemer er laget som generelt laboratorieutstyr hvor man skal kunne
dekomponere mange slags prøver under svært
forskjellige betingelser. Sett i lys av at man arbeider med
konsentrerte syer under høyt trykk og høy
temperatur, er sikkerhetssystemene både for ovn og for
beholdere svært viktig.
Foredraget vil ta for seg kjemiske problemstillinger knyttet til bruk
av inkompatible blandinger av forskjellige syrer, og problemstillinger
rundt bruk av mikrobølger som direkte
energioverføring til prøver som har eksoterme
reaksjoner.
Videre vil foredraget presentere forskjellige sikkerhetssystemer
knyttet til ovner og beholdere både når det gjelder
trykk og temperatur.
Det vil også bli presentert eksempler på
dekomponeringer hvor reaksjonene har vært så
hurtige at de første sikkerhetsbarrierene ikke har greid
å fange opp reaksjonen. Det vil bli gitt eksempler
på reaksjoner som er uheldige i mikrobølgeovn.
Bestemmelse av biomarkører er et viktig redskap ved diagnose av sykdommer. Biomarkører er kroppsegne forbindelser, ofte proteiner, hvor utskillingsgraden endres ved patologiske tilstander. Å bestemme konsentrasjonen av gitte markører i blod eller urin kan brukes til å stille en presis diagnose, gi prognoser for utfall av sykdom, eller monitorere sykdomsutvikling eller legemiddeleffekter. I dag gjøres biomarkørbestemmelse hovedsakelig ved hjelp av immunologisk baserte teknikker som ELISA. Disse er enkle i bruk, relativt billige og veldig sensitive. Ulempen er at teknikkene har releativt lav spesifisitet noe som kan føre til feilaktige diagnoser.
Det utføres mye forskning for å bedre
biomarkørbestemmelsen slik at man kan stille bedre og mer
presise diagnoser. LC-MS er en teknikk med høy spesifisitet,
men
det har vist seg å være vanskelig å
oppnå samme
sensitiviteten som ved bruk av immunobaserte teknikkene. Ved
å kombinere immunoaffenitetsopprensing med LC-MS/MS kan man
dra
nytte av både den høye sensitiviteten av
immunobaserte
teknikker og den høye spesifisiteten av LC-MS/MS, noe som
igjen
vil kunne føre til mer presise diagnoser.
Potensialet av
kombinasjonen vil bli vist ved bruk av teknikken på reelle
problemstillinger på kjente kreftmarkører. Analyse
av
pasientprøver vil også bli vist.
AbstractDet fines mange gode grunner til å vite helt nøyaktig hvordan et legemiddel fungerer. Måten et legemiddel ofte fungerer på, er at den binder seg til et protein, slik at proteinet ikke er lengre like aktivt. For eksempel, ibuprofen binder seg til COX, som er et enzym som starter en rekke med reaksjoner som gjør at vi for eksempel føler smerte eller får feber. Hvilket protein et legemiddel binder seg, og hvordan den binder seg, kaller man ofte en ”mode of action” (MOM).
Det å kjenne legemiddelets MOM gir mange fordeler. For eksempel, det gir større trygghet i bruk av medisinen, man kan lage bedre medisiner når man vet hva målproteinet er, og det kan gi oss et større innblikk i proteinets rolle i en sykdom. Men for mange legemidler er det vanskelig å bestemme MOM. Dette er fordi det er titusensvis av proteiner i en celle, og i prinsippet kan alle proteiner være mål.
Det finnes mange forskjellige metoder for å bestemme MOM [1]. Felles for mange er at man tilsetter et legemiddel til en prøve (for eksempel en cellelysat), og fisker ut legemiddelet med proteinet hengende på. Det høres enkelt ut, men dette er en stor teknisk utfordring. For eksempel, stoffet kunne festes i et ”snøre” (for eksempel en biotin gruppe) for å kunne fiskes ut (for eksempel med en kule med streptavidin). Da må man ofte derivatisere stoffet eller syntesere et analog. Dette kan både være tidskrevende, og stoffets aktivitet kan svekkes. I tillegg må man ha kontroll på hvilke proteiner i en celle som binder seg ikke-spesifikt til legemiddelet/kulen (for eksempel med ioniske eller hydrofobiske interaksjoner).
Med andre ord, er bestemmelse av MOM ofte en skikkelig
analysisk
utfordring. På Universitetet i Oslo, holder vi på
å
utvikle en ny metode for bestemmelse av MOM, hvor vi håper
å kunne slippe å derivatisere eller resyntesere
antikreftlegemidler som vi utvikler innen CAST (Cancer Stem Cell
Research Center) [2, 3]. Vår metode krever gode
kromatografiske
metoder for høyoppløselige separasjoner av
intakte
proteiner. Vi kombinerer slike separasjoner fluorescens polarisasjon
(FP) [4] og LC-MS [5]. I mitt foredrag vil jeg vil diskutere siste nytt
i vår metodeutvikling.
Rene, enkle kjemiske modellsystemer kan være vanskelige nok å tenke på og tale om. Likevel, i praksis arbeider vi kjemikere som oftest med svært sammensatte systemer, - enten vi analyserer mat, miljø eller mennesker.
Prøvene våre kan inneholde et høyt antall kjemiske stoffer, mange av dem relativt like, mange av dem med flere mulige molekylære tilstander, mange av dem ukjente for oss, og mange av dem med mulig biologisk eller økonomisk betydning. Måleteknikkene har fått bedre og bedre oppløsning, slik at vi i dag kan detektere og kvantifisere og identifisere (?) stadig flere stoffer i stadig lavere konsentrasjoner. Både i kromatografi, i spektrometri og ulike kombinasjonsteknikker kan vi vanligvis få ut data for et stort antall mer eller mindre rene ”topper” - om vi vil. Etter hvert som billed-dannende teknikker utvikles, vil vi kunne ta vare på naturlig heterogenitet i prøvene og unngå artefakter.
Men jo mer data, jo mindre informasjon – med mindre vi har verktøy og kompetanse til å tolke dataene. Dessuten er ikke alle ”toppene” relevante for vår problemstilling. Problemet er bare at vi ikke på forhånd kan vite hvilke ”topper” som er relevante og hvilke som ikke er det.
Den tradisjonelle måten å unngå mental forvirring er å redusere antallet topper man må ta hensyn til – for eksempel ved måten man setter analysene på, eller simpelthen ved å ignorere alle topper som ikke inngår i ens aktuelle arbeidshypotese eller i ens generelle fagtradisjon, eller som man ikke kjenner navnet på. I praksis må alle gjøre dette i noen grad, men overdrives denne forenklingen, risikerer vi å miste viktig informasjon, og vår evne til problemløsning reduseres.
En annen og bedre måte å beholde oversikten på er å ta i bruk data-analytisk programvare. Dette vil forenkle og fokusere informasjonen i de store datatabellene fra måleinstrumentene. Det kan inngå både instrument-spesifikk forbehandling av rådata, og mer generiske teknikker for selektivitets-forbedring, statistiske hypotesetesting og ren eksplorativ analyse. Noe data-analytisk metodikk blir ofte levert med dagens instrumentering i analytisk kjemi, og kan brukes som mer eller mindre ”black box”. Men har man livsmot, kan det lønne seg å skaffe seg mer generisk data-analytisk forståelse og programvare, ikke minst hvis man vil kombinere data fra flere ulike typer instrumenter på de samme prøvene. Tradisjonell statistisk hypotese-testing er vel og bra, men den minst spennende delen av dataanalysen! I kjemiens år må vi bi bedre til å lytte til den virkelige verden, ikke bare til våre egne teorier om den.
Fagfeltet kjemometri har utviklet en rekke datamodelleringsteknikker som hjelper brukere til å øke informasjonsinnholdet i kjemiske måledata. En typisk anvendelse er multivariat kalibrering, der man øker selektiviteten i mangekanals måledata ved matematisk ”opprensning”, slik at behovet for fysiske og kjemisk opprensning av prøver før måling reduseres. Dette kan gi drastisk økning i analysekapasiteten. En annen typisk anvendelse er å bruke en matematisk selv-dekomponering av data til å få oversikt over sammenhengene innen og mellom store datatabeller. Brukere av slike programmer får stadige kick av å oppdage uventede, men systematiske mønstre i egne og andre data. Norsk og nordisk kjemometri har vært og er blant verdens ledende i feltet. Det er faktisk både enkelt og morsomt, men det må selvfølgelig læres, som alt annet.
Kjemometrien er utviklet av og for kjemikere, men med sterk støtte fra statistikere og matematikere. Pussig nok kan man bruke samme statistiske teknikker og samme programvare til både instrumentkalibrering, hypotesetesting, prøveklassifikasjon og eksplorativ oversikts-analyse. Det gjør livet enklere når man som kjemiker ønsker å bruke sin tid på kjemi, og ikke på statistikk.
Kjemometrien er i bruk innen mange typer analytisk
kjemi, både organisk og uorganisk. Moderne biospektroskopi
kan tjene som modell for hva som kan oppnås når
kjemometri og analytisk kjemi kombineres. Men også
metabolomikk og andre moderne kjemiske
”-omics” typer - utviklet for
å unngå overforenkling og i stedet vise respekt for
livets kompleksitet - egner seg godt for kjemometrisk data-modellering.
Bruken av kjemometri som ”FUGE-masse” innen moderne
funsjonell genomikk og biologi på Campus Ås vil bli
brukt som illustrasjon.
Spyttprøver blir i økende grad brukt til
analyse av
rusmidler for eksempel til forskningsprosjekter for kartlegging av
rusmiddelbruk, rusmiddelkontroll i yrkeslivet, screening av
bilførere og oppfølging av rusmiddelavhengige
pasienter
som deltar i legemiddelassistert rehabilitering. En stor fordel med
spytt som medium er at prøvetakingen er rask og enkel, og
kan
gjøres under oppsyn uten at det oppleves som krenkende for
prøvegiver.
På 1990-tallet var det store forventninger til bruk av
spyttprøver for rusmiddeltesting og analyse av legemidler
for
terapikontroll. Mange forskere antok at man ville kunne estimere
konsentrasjonen av et legemiddel eller rusmiddel i blod basert
på
analyse av spytt dersom man fikk beregnet ratioene mellom
konsentrasjoner i spytt og blod ved kontrollerte
prøvetakingsbetingelser. Senere forskning har imidlertid
vist at
dette ikke er gjennomførbart på grunn av store
inter- og
intra-individuelle forskjeller i ratioene mellom spytt og blod. I de
senere år har det vært større fokus
på bruk
til rusmiddelkontroll som en erstatning for urin, og flere studier har
funnet god kvalitativ overensstemmelse.
Spytt kan analyseres både med hurtigtester og immunologiske
eller
kromatografiske laboratoriemetoder. Lite prøvevolum og til
dels
svært lave konsentrasjoner for enkelte stoffer, for eksempel
THC
og benzodiazepiner, gir en del analytiske utfordringer. Foredraget vil
diskutere fordeler og ulemper med rusmiddeltesting i spytt.
Abstract missing
Hvorfor screening av fremmedstoffer i miljøet?
På den ene siden har samfunnet et legitimt ønske om et giftfritt miljø. På den andre siden så påvirker all menneskelig aktivitet vårt miljø og de færreste ønsker å gå glipp av alle de goder som det moderne industrisamfunnet tilbyr: Legemidler kan skilles ut gjennom urin og kommer via avløpsvann ut til elver, innsjøer og fjorder. Allværsjakker inneholder perfluorerte forbindelser som kan spores til og med i Arktis. Og selv god gammel vedfyring bidrar med dioksinutslipp til luft. Det betyr at vi trolig må leve med at vi utsettes for fremmedstoffer, men forekomst må begrenses slik at de ikke medfører fare for uønskete helse- og miljøeffekter. Det er derfor nødvendig at man hele tiden holder kontroll med utslipp og forekomst i miljøet.
Siden ressursene for overvåking er begrenset kan bare de viktigste fremmedstoffer inngå i en kontinuerlig overvåkning. Det store flertallet av mulige fremmedstoffer vet man lite om og kun en stikkprøveaktig overvåkning eller screening er mulig. Ved screeningen ønsker man å få et oversiktsbilde over en større stoffgruppe og sortere ut stoffer som muligens kan være problematiske.
Utfordringene med screening er mangfoldige og begynner ved prioritering av stoffer og utvalg av medier man undersøker, hvor i landet og når man tar prøver, om man tar mange forskjellige prøvetyper eller heller et stort antall like prøver men fra forskjellige steder. For oss kjemiker er det kanskje mest spennende hvordan man påviser og kvantifiserer stoffene i de forskjellige miljøprøver. Og til slutt hva betyr egentlig resultatet, det vil si en sammenfattende vurdering som veier funnene i miljøet opp mot det man vet om stoffenes miljø- og helseeffekter.
Avslutningsvis gis det et tilbakeblikk på resultater
på
mer enn 10-års screening av fremmedstoffer i
miljøet.
Statens arbeidsmiljøinstitutt har i perioden 2007-2009 gjennomført en undersøkelse av arbeidsmiljøet under arbeid med smøring av ski blant profesjonelle skismørere fra flere nasjoner i konkurransesituasjon i nordiske grener og skiskyting arrangert i Norge.
Studien har omfattet medisinske undersøkelser (lungefunksjonsmålinger), målinger av biomarkører i blod, personlige eksponeringsmålinger av kjemiske forurensinger i skismørernes innåndingssone, samt undersøkelser i modellforsøk av de enkelte arbeidsoppgavers betydning for eksponering.
Denne presentasjonen vil ta for seg hva moderne
skismøring
består av, resultatene av kartleggingen av det kjemiske
arbeidsmiljøet til profesjonelle skismørere samt
hvilke
analytiske metoder som har blitt benyttet til å karakterisere
det
kjemiske arbeidsmiljøet.
AbstractModerne måleteknikk genererer så mye rådata at vi alle er blitt avhengige av matematisk og statistisk data-behandling, bevisst eller ubevisst. Selv et tilsynelatende enkelt pH-meter inneholder ganske avanserte matematiske transformasjoner av rådata. Og moderne flerkanals måleinstrumenter, som spektrofotometre, konfokale mikroskoper, GC-MS og LC-MS instrumenter, DNA-sekvenseringsinstrumenter osv gir fra seg tusener av ”analyser” – målevariabler – per prøve. Så vi generer fort svære data-tabeller, som trenger matematisk og statistisk behandling for å gi mening. Slike datasett kan noen ganger være bare tull, men med fornuftig forsøksplanlegging av forsøk, og fornuftig forbehandling av rådata, blir de svært informative.
Data må alltid tolkes i lys av fagfolks bakgrunnskunnskap, enten det gjelder kjemiske profileringer eller sensoriske paneldata og forbrukerundersøkelser. Og mange slags data må kombineres, om man vil forstå matens komplekse kvalitetsvariasjoner. Spørsmålet er hvordan vi kan jobbe effektivt, ha det morsomt og unngå fremmedgjøring og feiltolkning.
Kjemometrien er utviklet av og for kjemikere, men med sterk støtte fra statistikere og matematikere. Man kan bruke samme statistiske teknikker og samme programvare til både instrumentkalibrering, hypotesetesting, prøveklassifikasjon og eksplorativ oversikts-analyse. Dette faktum gjør livet enklere, siden de fleste kjemikere ønsker å bruke sin tid på kjemi, og ikke på statistikk. Fagfeltet kjemometri har - sammen med søsterfeltet sensometri - utviklet en rekke datamodelleringsteknikker og programpakker som hjelper brukere til å øke informasjonsinnholdet i kjemiske og sensoriske måledata. Brukerne av slike verktøy bør sette av tid til å forstå prinsippene som brukes i programmene, men behøver slett ikke bli spesialister i statistikk.
En typisk anvendelse er multivariat kalibrering, der man
øker selektiviteten i mangekanals måledata ved
matematisk ”opprensning”, slik at behovet for
fysiske og kjemisk opprensning av prøver før
måling reduseres. Dette kan gi drastisk
økning i analyse-kapasiteten. En annen
typisk anvendelse er å bruke en eller annen type matematisk
selv-dekomponering av data til å få oversikt over
sammenhengene innen og mellom store datatabeller. Brukere av
slike programmer får stadige kick av å oppdage
uventede, men systematiske mønstre i egne og andre data.
Norsk og nordisk kjemometri og sensometri har vært og er
blant verdens ledende, og næringsmiddel-sektoren er en viktig
arena for nytenkning i disse feltene: Både maten og mennesket
er nemlig så komplisert at det holder ikke å drive
hypotesetesting på ett og ett stoff, eller en og en
egenskap.
Moderne full-spektrums biospektroskopi kan tjene som modell for hva som kan oppnås når multivariat analyse og analytisk kjemi kombineres. Måler man høydimensjonelle infrarøde og nær-infrarøde spektra fra mange relevant prøver, har man ofte et sant overskudd av informasjon. Derfor har man muligheten til å gjøre mange ulike kvantitative bestemmelser fra samme sett av måledata. Man kan også oppdage nye problemstillinger.
Mange andre analysetyper egner seg også godt for
kjemometrisk datamodellering, som høydimensjonell
metabolomikk og andre moderne multirespons ”-omics”
typer, utviklet for å unngå overforenkling og i
stedet vise respekt for livets kompleksitet. Men spesielt er
kjemometri og sensometri viktig når man vil sammenholde
kvantitativ informasjon fra flere fagfelt.
Genopvarmet kød smager hengemt, især retter lavet af gris eller kylling. Det er fedtstoffet, der iltes hjulpet på vej af jern frigjort som jern-ioner fra jernholdige proteiner ved opvarmning. Sådan katalytisk lipidoxidation danner flygtige forbindelser ved at kløve de umættede lipider. Jern er et eksempel på en prooxidant i vore fødevarer. Jern er nødvendigt for os, men også en trussel, idet det igangsætter dannelse af frie radikaler. Nye anbefalinger fra kræftforskerne siger højst 300 g rødt kød om ugen (!) og slet ingen forarbejdede kødprodukter. Kød er rødt, fordi jern er bundet i hæm-proteiner, der bærer oxygen rundt. Under fordøjelse bliver disse proteiner som for eksempel myoglobin aktivt og bombarderer væv i tarmen med aggressive frie radikaler. Heldigvis kan vi sammen med oksekødet spise grøntsager eller krydderier spækket med polyphenoler, der kan indfange de frie radikaler og dermed virke som antioxidanter.
Mælk fra kølediskens stærke lys kan lugte som en hårlok strejfet af stearinlysets flamme. Det er proteiner, der nedbrydes oxidativt, men nu med lysaktiveret B-2-vitamin som prooxidant. Dette vitamin (riboflavin) er gult og er ekstremt oxiderende efter absorption af lys, men gendanner grundtilstanden efter reaktion. Denne type prooxidanter kaldes fotosensibilisatorer. Planternes grønne chlorophyll er et andet eksempel, som kan beskadige den frie olivenolie hensat i køkkenvinduets lys. Mælkens svedne lugt skyldes riboflavins oxidation af aminosyren methionin til små letflygtige svovlforbindelser. Mælken får senere metalsmag, idet lysaktiveret riboflavin efter proteinernes aminosyrer angriber fedtstofferne. Lysaktiveret riboflavin kan deaktiveres af vitamin E og vitamin C og af flavonoider, som de findes i bær og frugter. Disse stoffer bliver dermed antioxidanter og beskytter mod lysbeskadigelser. Mælks indhold af andre forbindelser, der beskytter mod lysbeskadigelse, kan fremmes gennem fodervalg. Urinsyre vil således indirekte beskytte den vigtige folsyre mod lysnedbrydning ved at reagere hurtigere med aktiveret riboflavin end folsyre.
Enzymer kan også være prooxidnater. Mælk er det perfekte næringsmedium for bakterier. Mælk indeholder derfor tre enzymer, der trinvist aktivere oxygen for at dræbe bakterier. Det molybdæn- holdige xanthinoxidase giver først superoxid, der dernæst omdannes af kobber/zink-enzymet superoxiddismutase til hydrogenperoxid, som det jernholdige laktoperoxidase bruger som oxidator, så thiocyanat kan bliver til thiocyanogen og andet, bakterier ikke tåler. Denne aktivering af oxygen kan løbe løbsk så mælkens proteiner og lipider tager skade, og enzymerne bliver derfor prooxidanter.
Evolution har balanceret på en knivsæg
mellem
prooxidanter nødvendige for oxygen-aktivering og
antioxidanter
som vigtig beskyttelse mod reaktive oxygen species (ROS), siden
fotosyntesen opstod i blågrønne alger for 3
milliader
år siden, og jordens atmosfære blev oxygenholdig.
Eksemplerne viser at denne balance genfindes i vore
fødevarer.
Læs mere i: Leif
H.
Skibsted: Understanding oxidation processes in food. In: Oxidation in
foods and beverages and antioxidant applications. Eds. Eric Decker,
Ryan Elias & D. J. McClements, Woodhead Publishing, Cambridge,
2010, 3-35. ISBN-978-1-84569-648-1
Antioksidanter er en betegnelse ofte brukt på molekyler som kan redusere oksidativt stress. Oksidativt stress oppstår ved at det dannes for mye reaktive biprodukter av oksygen som videre kan forårsake uønskede endringer i celler og vev. Et eksempel på dette er oksidering av DNA med påfølgende mutasjoner. Oksidativt stress er med andre ord en direkte konsekvens av at vi lever i en oksygenrik atmosfære. Imidlertid har evolusjonen sørget for utviklingen av et meget omfattende forsvarsverk (antioksidantforsvar) for å takle eller eliminere de toksiske bivirkningene av oksygen. Men det er også faktisk slik at systemer er blitt utviklet for å gjøre seg nytte av reaktive oksygenforbindelser, for eksempel til signaloverføringer i cellene eller til forsvar mot bakterier.
Antioksidanter kan også tilføres gjennom maten, særlig fra planter, og de mest kjente antioksidantene er vitamin C , vitamin E og karotenoider. I tillegg vet vi at det finnes tusenvis av andre molekyler med antioksidantliknende egenskaper som er mer eller mindre karakterisert. Det er vist i en rekke studier på mennesker, dyr og cellekulturer at plantebaserte næringsstoffer har gunstige effekter på sykdomsprosesser hvor oksidativt stress er en komponent. En logisk slutning har da vært å tilskrive effekten av plantebasert kosthold på evnen til å redusere oksidativt stress ved hjelp av antioksidanter. En hypotese har vært at antioksidantene direkte kan reagere med skadelige oksygenforbindelser. I de senere årene har dette bildet blitt noe mer nyansert. Det har vist seg at mange av de stoffene man kjenner som antioksidanter også har mange andre egenskaper som ikke er direkte relatert til deres antioksidantegenskaper. I tillegg har store kliniske intervensjonsstudier med antioksidantsupplementer gitt uventede resultater.
I foredraget vil det bli diskutert hva antioksidanter er, om
de er nødvendige og hvilke roller disse har i ulike
biologiske prosesser og sykdomsutvikling.
AbstractKjøtt er det mest næringsrike vi kan spise; det gir essensielle aminosyrer, bioaktive stoffer, vitaminer og mineraler. Kjøtt er rikt på oljesyre og fettsyrer med biologiske virkninger, som CLA (i drøvtyggerkjøtt), essensielle fettsyrer i omega-6 og omega-3 familiene; arakidonsyre, EPA, DPA and DHA.
Kjøtt har forbedringspotensial, og vi mener at husdyrprodukter bør være så sunne som mulig. Kjøtt, og spesielt kjøtt fra kylling og svin kan bli sunnere å spise hvis innholdet av omega-6 fettsyrer reduseres og innholdet av omega-3 fettsyrene økes, samtidig som innholdet av blant annet sporstoffet selen økes.
Økonomiske merkostnader til å optimalisere kjøttsammensetningen vil trolig være beskjedne i forhold til kostnadene ved dårlig folkehelse. Globale økologiske hensyn kan kanskje tale for å minske totalkonsumet av husdyrprodukter, men det som folk uansett spiser av husdyrprodukter bør ha en helsemessig mest mulig gunstig sammensetning. Ved å korrigere husdyrproduktenes sammensetning vil det helsemessig ikke bety noen vesentlig forskjell om folk foretrekker å spise kjøtt eller fisk. Fisk er rik på selen og flere viktige mineraler og omega-3 fettsyrer, men fisk er en begrenset ressurs i verden.
Kjøtt, innmat og egg er kildene for arakidonsyre i kosten. Arakidonsyre omdannes i cellene våre til hormonlignende stoffer. Det dannes også hormonlignende stoffer fra omega-3 fettsyren EPA. Disse to fettsyrene ligner hverandre i struktur, og det er en konkurranse mellom dem i cellene . De omdannes til stoffer med delvis forskjellig virkning, og produksjonshastigheten kan være meget forskjellig. Vi trenger en balanse mellom disse stoffene samt at det verken bør dannes for lite eller for mye. I vestlig kosthold er det for høyt inntak av omega-6 fettsyrer sammenlignet med inntaket av omega-3 fettsyrene, hvilket fører til både overproduksjon og ubalanse.
Omega-6 fettsyrene i kostholdet kommer fra de viktigste planteoljene som brukes her til lands, slik som soyaolje, solsikkeolje, maisolje og margarin og fra kjøtt fra dyr som er fôret med kraftfôr. Kraftfôr er laget av blant annet korn og soyaolje, og det er mye mer omega-6 enn omega-3 fettsyrer i disse råvarene.
Omega-3 fettsyrer får vi fra linfrøolje, rapsolje og fisk. Gras og grønne blader inneholder også omega-3 fettsyrer, og dyr som er fôret med grovfôr har mer omega-3 fettsyrer i kjøttet. Omega-3 fettsyrene EPA og DHA kalles ofte ”fiskefettsyrer”, men vi får en betydelig andel av disse fettsyrene fra kjøtt fra grasfôrede dyr. På Island og i Frankrike bidrar kjøtt mer enn fisk til inntaket av disse omega-3 fettsyrene i kostholdet. Derfor; kjøtt kan være en viktig kilde for EPA og DHA.
Ved Institutt for husdyr- og akvakulturvitenskap, UMB har vi byttet ut tilskuddet av soyaolje i kraftfôret med rapsolje og linolje i fôr til kyllinger, og har derved fått redusert innholdet av arakidonsyre i kyllingkjøttet og økt innholdet av EPA.
Innholdet av selen i kyllingkjøtt er økt
opp til samme
nivå som seleninnholdet i fisk ved å tilsette
organisk
selen (0,84 mg/kg) til kraftfôret .
Konklusjon
Referanser
Fiskeoljer inneholder en høy andel av de langkjedede flerumettede omega-3 fettsyrene (blant annet EPA (20:5n-3) og DHA (22:6n-3)) som er vist å ha positive effekter på menneskers helse. Folk blir stadig mer oppmerksomme på betydningen av å sikre seg tilstrekkelig inntak av omega-3 fettsyrer. I tillegg til fet fisk er opprensede fiskeoljer en god kilde for disse fettsyrene. Siden marine lipider inneholder en høy andel av de flerumettede omega-3 fettsyrene, så er de meget utsatt for harskning. Harskning er en prosess som skjer når olje kommer i kontakt med oksygen i nærvær av ulike katalysatorer (metaller og andre prooksidanter). Denne reaksjonen fører til dannelse av primære harskningsprodukter, vanligvis kvantifisert som peroksidverdi.. Disse primære harskningsproduktene som ikke lukter eller smaker spaltes videre til sekundære harskningsprodukter som er forbindelser med lavere molekylvekt og som er ansvarlige for uønsket harskt lukt og smak i olje. For å hindre harskning av marine lipider er det derfor viktig at lipider lagres under oksygenfrie betingelser, prooksidanter fjernes eller inaktiveres og at de riktige antioksidantene tilsettes.
Vi har studert effekt av forskjellige prooksidanter som jern og methemoglobin på oksidasjon av marine fettsyrer og har konkludert ut fra våre studier at det er forskjellige oksidasjonsmekanismer som er involvert i oksidasjon katalysert av henholdsvis jern og methemoglobin. Methemoglobin er en langt sterkere prooksidant enn jern. Både jern og methemoglobinkatalysert oksidasjon kan hindres ved reduksjon av lagringstemperaturen til marine oljer. Oksidasjonshastigheten kan også reduseres ved valg av riktig pH. Oksidasjon går raskest når pH i olje/vann systemet er ca 5.5. Kelatorer (som EDTA) kan binde jern og hindre oksidasjon ved pH der kelatorene er negativt ladet. Proteiner kan også bidra til å hindre oksidasjon, men pH i oksidasjonsystemet bør også her være slik at proteinene har netto negativ ladning. Valg av antioksidanter er svært viktig og må velges avhengig av prooksidantene i systemet. For eksempel vil kaffeinsyre fungere som prooksidant ved jernmediert oksidasjon, men som antioksidant ved methemoglobin mediert oksidasjon.
I april 1953 publiserte tidsskriftet Nature en artikkel av
James Watson og Francis Crick. Artikkelen viste seg å
inneholde løsningen på hvordan DNA-molekylet var
oppbygd. Foredraget vil ta for seg den unge norske kjemikeren Sven
Furbergs bidrag til klarleggingen DNA-molekylets oppbygning. Furberg
oppholdt seg i årene 1947-1949 ved John Desmond Bernals
laboratorium ved Birkbeck College ved University of London, og gjorde
der et arbeid som hadde stor betydning for videre analyse av
DNA. I den omfangsrike litteraturen om hvordan gåten ble
løst spiller imidlertid Furberg - med et par unntak - en
marginal rolle. Jeg vil diskutere nærmere hva hans bidrag
besto i, og hvilken rolle bidraget spilte i andre forskeres diskusjoner
av problemet. Viktige forskere i denne sammenhengen blir
særlig - foruten hovedpersonene Watson og Crick ved
Cavendish-laboratoriet i Cambridge - Rosalind Franklin og Maurice
Wilkins ved King's College i London. Jeg vil også se
på hvilken rolle karakteristika ved de ulike laboratoriene
som var involvert kan ha spilt for den virkningen ulike bidrag til
løsningen fikk. På 1970-tallet ble læreplanene endret slik
at kjemi ble
et ordentlig undervisningsfag i videregående skole. Verken
lektorene i det daværende gymnas eller Skolene var
forberedt på dette. Det var nok dette som
førte til
at Skolelaboratoriet for kjemi ble opprettet. Skolelaboratoriet
for kjemi startet virksomheten høsten 1974 med
Einar Wang
Lund som leder. Det skulle virke som sentrum for kursvirksomhet og
etterutdanning, og det er det fortsatt. Helt uventet
dør
Einar Wang Lund i august 1977, og spørsmålet
dukker opp om
hva instituttet skal prioritere ved nytilsettelse. Truls
Grønneberg, som overtok som leder av Skolelaboratoriet 1980,
valgte å prioritere det skolerettede arbeidet som han vil
fortelle mer om. Men når han nå
går av til
neste år etter å ha ledet
Skolalboratoriet i mer enn
30 år, er spørsmålet dukket opp igjen:
Hva vil
instituttet prioritere ved en eventuell nytilsettelse?H - Kjemiens historie
H1
Furbergs bidrag til strukturen av DNA.
Edgeir
Benum
Seksjon for historie, Institutt for arkeologi, konservering
og historie, Universitetet i Oslo
H2
Skolelaboratoriet for kjemi UiO 30 år
Truls
Grønneberg
Kjemisk institutt, Universitetet i Oslo
H3
Ble nye navn på H, N og O i 1950-årene en
suksess?
Brit
Skaugrud
Hvor kom de gamle navnene vannstoff, kvelstoff og surstoff fra, og
hvorfor ble disse navnene endret midt på 1950 tallet? Etter at
nomenklaturutvalget i Norsk kjemisk selskap i 1962 publiserte en liste
over grunnstoffnavn, ble de nye navnene hydrogen, nitrogen og oksygen
(og karbon) tatt i bruk i ordbøker og nye
lærebøker. En enkel studie av oppslagord i Aftenposten fra
1950 til i dag, viser at selv om vi alle lærte de nye navnene i
skolen, lærte ikke alle å bruke dem. Studien viser
også at det tok mer enn 15 år før oksygen ble brukt
oftere enn surstoff i avisen, mens nitrogen ble brukt like ofte som
kvelstoff allerede i 1962.
Det
første NMR-spektrometeret i Norge ble
installert på Sentralinstitutt for industriell forskning (SI)
i 1960. Det var
tilgjengelig for alle i Norge slik annet utstyr på SI var. SI
ble etablert av
forskningsrådet (NTNF)
i 1949 og skulle
være et sted hvor norsk industri kunne få adgang
til moderne utstyr og forskere
med kompetanse. Staben var bygget opp i 1950-årene og flyttet
inn i et nybygget
hus på Gaustad nær universitet på
Blindern i 1956. I 1993 fusjonerte SI med
SINTEF.
Abstract missing Abstract missing Despite bearing a potentially reactive site on the central
backbone carbon, aryl substituted β-diketiminate ligands are
widely employed to stabilize unusual oxidation states and coordination
environments and to support active catalytic species. The
five-coordinate Pt(IV) complex, (tBuMe2nacanc)PtMe3 (tBuMe2nacnac =
[((4-tBu-2,6-e2C6H2)¬NC(CH3))2CH]), undergoes a formal
cycloaddition reaction with molecular oxygen to generate a
six-coordinate species in which one of the oxygen atoms is bound to the
metal and the other is bound to the central carbon of the ligand
backbone. Over time this peroxo complex reacts further to generate a
product in which the oxygen-oxygen bond has been cleaved. The
cooperative reactivity between the central carbon of the ligand
backbone and the metal with molecular oxygen and other unsaturated
species will be discussed. Much of the effort spent on optimization of Grubbs
ruthenium-based catalysts for olefin metathesis has been focused on
variation of the donor ligands, both the ones leaving to generate the
active 14-electron alkylidene LCl2Ru=CHR and the remaining neutral
ligands L. Previous studies have identified L to be among the most
important activity-promoting properties. In recent years, novel
amido-like, neutral ligands with donor strengths comparable to or
greater than that of N-heterocyclic carbenes have been developed.
Unlike dative ligands so far employed in olefin metathesis, the new
nitrogenous ligands are both good [sigma] and [pi]-donors. These
compounds thus appear to have design elements that could prove useful
for the remaining ligands L, but may, in some complexes, also be
efficient dissociating ligands. We have embarked on an investigation of
these compounds as ligands in ruthenium-based olefin metathesis
catalysts. Here, we report the reaction of 2-isopropyl
β-carboline with the first and second generation Grubbs
catalysts to give the corresponding mono-substituted complexes in good
yield. Both compounds show high olefin metathesis activity at room
temperature. Theoretical investigations have also been employed to
elucidate the nature of the β-carboline ligand in these
complexes. Adsorption microcalorimetry is the most reliable method for
the determination of the energy of the bonds between the adsorbed
species and the catalyst surface, information that is useful in
understanding the nature of the catalyst and essential in the detailed
(microkinetic) modelling of catalytic reactions. Although supported
cobalt catalysts are used commercially in the Fischer –
Tropsch synthesis (FTS), there is limited knowledge available regarding
the surface adsorption energetics (heat of adsorption). Most of the
studies dealing with the adsorption heat of different molecules on
cobalt are based on theoretical calculations or indirect experimental
determination, e.g. temperature programmed desorption (TPD). In this
study a calorimetric setup for the accurate and direct determination of
the heat of adsorption (ΔHads) as a function of adsorbed
amount is presented. A series of cobalt catalysts with constant cobalt
loading and a range of cobalt particle sizes is investigated. Over the
range of cobalt particle sizes investigated (4-15 nm) we do not observe
any significant difference in the heat of adsorption of hydrogen. This
is quite surprising, since smaller particles (< 6-8 nm) expose a
larger fraction coordinatively unsaturated sites, and also have very
different catalytic properties compared to larger particles. The delamination of layered zeolite precursor PREFER is
demonstrated under mild nonaqueous conditions using a mixture of
cetyltrimethylammonium bromide, tetrabutylammonium fluoride, and
tetrabutylammonium chloride in N,N–dimethylformamide (DMF) as
solvent. The delamination proceeds through a swollen material
intermediate which is characterized using powder X-ray diffraction
(PXRD). Subsequent addition of concentrated HCl at room temperature
leads to synthesis of UCB–2 via delamination of the swollen
PREFER material, and is characterized using PXRD, transmission electron
microscopy (TEM), and argon gas physisorption, which shows lack of
microporosity in UCB–2. 29Si MAS NMR spectroscopy indicates
lack of amorphization during delamination, as indicated by the entire
absence of Q2 resonances, and 27Al MAS NMR spectroscopy shows
exclusively tetrahedral aluminum in the framework following
delamination. The delamination process is reversible between
delaminated and swollen states, and requires both chloride and fluoride
anions in DMF. AbstractThe methanol to hydrocarbon (MTH) reaction is one of
the ways of improving the value of natural gas, biomass or coal. In
this paper the MTH reaction is studied over one-dimensional 10-ring
zeolites at various reaction conditions: ZSM-22 (TON), ZSM-23 (MTT),
ZSM-48 (MRE) and EU-1 (EUO). Temperatures between 350 and 500
°C and WHSV between 2 and 6 gg-1h-1 are investigated using a
fixed bed reactor. The products are analyzed using online GC. The
hydrocarbon species trapped in the channels of the material during the
reaction were liberated using the standard HF dissolution procedure and
analyzed using GC-MS. The catalysts converted comparable amounts of
methanol before complete deactivation at their optimum MTH condition.
Despite the small differences in the channel dimensions, the materials
displayed very different product spectrum. Except from EU-1, all the
catalysts gave high selectivity for hydrocarbons in the boiling range
of gasoline fuel, C5+ fraction. Unlike ZSM-22 and ZSM-23, EU-1 and
ZSM-48 catalysts displayed notable amounts of aromatics in their C5+
fraction, which are considered environmentally unfriendly. For ZSM-22,
ZSM-23 and EU-1 catalysts the deposition of coke within the channels do
not affect the selectivity, instead, the change in selectivity with
reaction time can be regarded as change in contact time. The
involvement of 12-ring side pocket of EU-1 zeolites for the MTH
reaction is revealed both by the unexpected catalytic behavior and
analysis of retained species within the side pocket. channel dimensions
between ZSM-22 and ZSM-23 zeolites is revealed by coke analysis. This
contribution addresses the effect of small differences in channel
system of one dimensional 10 ring zeolite on stability and selectivity
during the MTH reaction. Aggregation of silicon atoms in aluminophosphate structures
has been investigated, assuming that the process may be described as an
formation of a silica phase in form of shpere within the AlPO
structure. Using Ostwald ripening thermodynamics, as silicon atoms
aggregate to make bigger particles, the bulk energy increases as r3,
while the surface energy increases as r2. The surface term dominates
for small r and the bulk term at larger r. As the two energies have
different signs, a balance is reached at a given critical radius rc.
Critical radii have been estimated using bulk and interfacial energies
from molecular mechanics calculations. Surface energies of the
silicate/aluminophosphate interface were calculated using slab, rod,
and sphere models for interfaces between the phases of silica and
aluminophosphate compositions. We note that the energies vary very
little upon changing the curvature of the surface, and report
predictions with regard to the optimum silicon island size
distribution. Further, direct energy calculations were carried out for
different sizes of silicon islands in an AlPO matrix. Gratifyingly, the
same trend is found. Catalytic dehydrogenation of NGL compounds (ethane, propane,
butane) is an important industrial process. Although they have been
extensively studied, a complete understanding of reactions, including
activity, selectivity, coke formation and deactivation, on the catalyst
surface is still missing. We have prepared well controlled Pt
nanoparticles with different size and shape, which have different ratio
of surface sites and varied in the orientations of the surface such as
{211}, {111}, and {100}. Combining DFT simulation, the effects of
surface atoms and facets of Pt nanoparticles on the activity,
selectivity and intrinsic kinetics of propane dehydrogenation (PDH)
have been investigated for the first time. The oxychlorination reaction is a key step of PVC chemistry.
Copper supported on alumina is the base catalyst for this process. In
order to make this catalyst more selective and have long lifetime,
dopants are added.Effect of promoter is quite complex on the catalyst
can be described as it can influence on surface of catalyst and after
all on selectivty of the catalyst. So to understand basic mechanism of
promoter effect, we studied the doped catalysts using combined in situ
and in operando XANES/EXAFS, FTIR, UV-vis, CO chemisorption and
catalytic tests to elucidate the role that dopants (LiCl, KCl, CsCl,
MgCl2 LaCl3) have in the nature, relative fraction, reducibility and
dispersion of Cu-phases on CuCl2/γ-Al2O3
catalysts for C2H4. The doping eliminates all the surface Lewis acidity
in CsCl and KCl doped-catalysts and strongly suppresses it in the
remaining cases. The increase of the strength of the
Brønsted sites is remarkable in all cases but the CsCl doped
one. To understand both the effect of Cl- anions and dopant cations a
set of dopant free, HCl-impregnated and of Cu-free dopant-impregnated
supports have been investigated. Addition of chlorine decreases the
density and the strength of Lewis sites, while it increases those of
the Brønsted sites. Catalytic testing of each material
revealed that formation of chlorinated byproducts was directly
correlated with the density of Lewis acid sites. Furthermore, an This
work is important for the ethylene oxychlorination catalysis, as: (i)
the surface acidic sites of alumina are the main origin of undesired
side products, and (ii) the acidic sites play a key role in determining
the structure and reactivity of the supported active phase. The second
function is evidenced by the empirical correlation reported was found
between the strength of the Brønsted acid sites of the
support and the ν-(CO)
of CO adsorbed on the reduced fraction of the active copper chloride
phase. The present study is aimed to complement the published
literature on alumina, underlining the usefulness of the molecular
approach made by IR spectroscopy low temperature adsorbed CO to
investigate the surface of catalyst support. In Conclusion, all metal
dopants had a significant effect on controlling the byproducts
formation compared to the base catalyst. In this study, we evaluated the effect of gallium concentration on
the performance of Rh promoted perovskites during partial oxidation of
methane at 673K. Ga modified perovskite-type oxides
La0.75Sr0.25Fe0.8(1-x)Co0.2(1-x)GaxO3-δ (x=0.1, 0.25, 0.4, 0.6)
were prepared by the nitrate-citrate method and annealed at 1000oC in
air. The synthesized samples were single perovskite phase materials,
with a small amount of secondary LaSrGa3O7 phase in sample with 60 %
gallium only. The initial methane conversions of Rh promoted Ga
modified perovskites are different. This is probably due to different
metallic dispersion in the catalysts. The catalytic tests of Rh
promoted perovskites with Ga concentration 10 and 25 % show a higher
average activity for partial oxidation of CH4 to synthesis gas compared
to unmodified oxides. The average selectivity to CO yield increases
with Ga concentration, except for the catalyst with the highest gallium
concentration. The amount of CO2 produced during reaction significantly
decreases when Ga is introduced into the perovskites structure. The
catalytic tests showed that Rh promoted Ga modified perovskites
catalysts exhibit a more rapid deactivation with increasing amount of
gallium. This is probably due to carbon deposition on the catalysts
surface.
Ka - Katalyse
Ka1
Alumina Supported Co Catalysts for Fischer-Tropsch Synthesis.
Effect of Particle Size and Alumina Modifications on Activity and
Selectivity.
Anders
Holmen
NTNU
Ka2
Chemistry for the Conversion of CO2 .
Richard
H. Heyn
SINTEF
Ka3
Metal-Ligand Cooperativity in O2 Activation: Observation of a
"Pt–O–O–C" Peroxo Intermediate.
Margaret
L. Scheuermann
University of Washington
Ka4
Ruthenium metathesis catalysts bearing 2-substituted
β-carboline ligands.
Fredrik
R. Hansen
University of Bergen
Ka5
Hydrogen adsorption on supported cobalt catalysts for the
Fischer-Tropsch synthesis studied by microcalorimetry.
Edd A.
Blekkan
NTNU
Ka6
Nonaqueous Fluoride/Chloride Anion-Promoted Delamination of
Layered Zeolite Precursors: Exfoliation of PREFER.
Einar
A. Eilertsen
University of Oslo
Ka7
Shape Selectivity in the Conversion of
Methanol-to-Hydrocarbon (MTH): the Catalytic Performance of
One-dimensional 10-ring Zeolites: ZSM-22, ZSM-23, ZSM-48 and EU-1.
Shewangizaw
Teketel
University of Oslo
Ka8
Silicon island in SAPO materials: Thermodynamic consideration
from atomistic modeling.
Mahsa
Zokaie
University of Oslo
Ka9
TitttelSelective C-H and C-C Bond Activation of Propane on
Platinum Nanoparticles with Different Sizes and Shapes.
Jun Zhu
NTNU
Ka10
FTIR, EXAFS and Kinteic studies on CuCl2
based catalyst for Oxychlorination of Ethylene.
Naresh
B. Muddada
University of Oslo
Ka11
Partial oxidation of methane to synthesis gas over Rh promoted perovskite based catalysts.
Radostina Palcheva and Marian Palcut
University of Oslo
The availability of multi-channel analytical techniques has
increased rapidly the last decade, especially in fields like
biospectroscopy, transcriptomics, proteomics and metabolomics. This
means that the same samples can easily be measured by several different
analytical techniques, giving a broad, but specific characterisation of
the samples. Each analytical technique gives rise to a data matrix, and
the objective is often to compare and combine information across
matrices. This leads to a need for targeted and powerful methods for
multivariate analysis of multiple data matrices.
Multiblock PLS (MB-PLS) is an established methodology for analysing
such data. It aims at modelling the general consensus pattern among
several data blocks, as well as how each block relates to the overall
pattern. However, MB-PLS does not give explicit quantitative
information about redundant variability in the X-blocks, and
interpretation can be tedious because there are many sets of
scores/loadings and it is not clear how each component represents the
various data matrices.
The new methodology that is presented here, Parallel and Orthogonalised
PLS regression (PO-PLS), aims at identifying redundant and unique
information in each data matrix. It splits the relevant information in
the predictor matrices into common components (present in two or more
matrices) and unique components (present in only one matrix). The
method allows for different number of predictive components from each
matrix, and explicitely states which components that are related to
which data matrices.
The PO-PLS methodology combines PLS regression and GCCA (Generalised
Canonical Correlation Analysis) to extract common components. It starts
by extracting components present in all matrices, and continues to go
through all relevant combinations sequentially. For each combination,
the current matrices are orthogonalised with resepect to the extracted
components. Finally, the unique components from each matrix are found
by PLS regressions of the orthogonalised matrices.
The methodology will here be illustrated by an example from
the food industry. Milk samples from a feeding experiment have been
measured with three spectroscopic instruments: Tensor 27 FT-IR
spectrometer from Bruker (dried milk samples), FT-IR Milkoscan
Combifoss 6500 from Foss (liquid milk samples) and a Kaiser Optical
Systems Raman RXN1 Analyzer (dried milk samples). In addition,
reference analyses of fatty acids have been made. The main objective is
to investigate the three spectroscopic techniques’ ability to
predict fatty acids. PO-PLS is used to determine if the spectra contain
overlapping or complementary information, and if it is beneficial to
combine different techniques in order to improve predictions.
With modern technologies scientists collect large amounts of data that are to be analysed and to be fitted with a model later on. Fitting a non-linear model to data is not a new problem, of course, and there exists a long list of methods for doing that. However, all these methods face various problems when fitting a model. Among such problems are: a long time of performance, ending up in a local optimum, a problem of choosing starting values and so on. Therefore, it has been of great importance to find such a method that would avoid as many existing problems as possible.
Here we present such a method suitable for data that represent some trend, which can be illustrated as curves (e.g., time series data). The method consists in having a data base of curves and a following direct look-up of a new curve in this "library". The data base in our example is a collection of curve sets from the 38 most common functions in science that can give monotonous increasing curves over an interval, from a straight lines to almost step-functions. For each function type the collection of curves is compressed by means of PCA and stored in a compact way as scores and loadings. A new curve is then projected onto the PCA model, and respective parameter values for each function model are obtained. At the end, a list of plausible functions along with their parameter estimates is available. The new method is non-iterative and does not depend on initial values of parameters. It has been tested on both artificial (with and without noise) and real data (from Two-Dimensional Gel Electrophoresis experiment). The results show that the direct look-up method gives at least as precise parameter estimation as traditional methods (e.g., hill-climbing) while speeding up the time of computation by a factor of about 24 compared to the simplex optimisation.
I multivariabel dataanalyse er formålet ofte å finne sammenhengen mellom to data-tabeller. Den typiske anvendelsen er å modellere konsentrasjon av kjemiske forbindelser eller kvalitet i et produkt fra mange instrumentelle målinger. Disse modellene benyttes deretter i en on-line implementasjon for prediksjon av produkt-kvalitet eller innhold av viktige ingredienser i prosessen. I andre situasjoner er formålet å etablere en modell for å identifisere eller klassifisere nye prøver. Eksempler er identifikasjon av råstoffer i farmasøytisk produksjon og test for å detektere at prøven ikke har blitt tilsatt andre kjemiske forbindelser. Fordi man ikke kan forvente selektivtet for enkelte signaler (variabler) i systemet benyttes multivariabel kalibrering for å inkludere/kompensere for kjente og ukjente interferenter. Disse modellene er empiriske av natur og man vet ikke i utgangspunktet hvilke variabler som gir den beste modellen. I mange anvendelser har man allikevel fra teorien informasjon om hvilke kjemiske strukturer som gir signal, f.eks. i FT-IR og NIR spektrospkopi. Den såkalte L-modellen muliggjør en direkte tolkning i form av en tredje datatabell som inkluderes i modellen; en "band assignment" tabell som beskriver den underliggende kjemi for det aktuelle måleprinsipp. Den tilhørende metoden for å anlaysere disse tre tabellen direkte har fått navnet L-PLS regresjon. Metodene har kan også benyttes inenfor bioinformatikk/genetikk/medisin der kjente uttrykk av enkelte gener eller kjemisk shift i NMR spektroskopi danner den tredje tabellen. Presentasjonen vil gå igjennom metoden på et konseptuelt nivå og eksempler på anvendelser vil bli vist.
In order to conduct statistical analysis of sets of two-dimensional
gel electrophoresis (2DE) images at pixel level instead of protein spot
level, a complete workflow enabling processing of 2DE images from
beginning (pre-processing) to end (statistical analysis) is needed.
This paper describes the first part of such a workflow, by introducing
two new preprocessing features: 1) A nonlinear image intensity
transformation to emphasize weaker protein signals. 2) A pixel-based
gel alignment method, which is based on a modified version of the
optical flow principle. This preprocessing is shown to simplify the
alignment between gels, and to reveal many more of the small protein
spots without loss of information concerning the big spots.
Spectroscopic techniques (UV, VIS, NIR, IR, fluorescence etc.)
are
increasingly used in food analysis, either for quality control or
scientific purposes. The techniques are popular because they are rapid
and can measure several key quality parameters. The last decade, the
opportunity to image these properties by the use of multi/hyper
spectral imaging has attracted attention. Chemical images are often
much more interesting than average concentrations. The approach enables
investigation of distribution and propagation of biochemical
properties, which is of significant value to the food science as well
as for on-line food control monitoring.
Different examples of multispectral imaging applied to foods are shown
in this presentation. On-line determination of fat and fat distribution
in salmon fillets, fat in meat trimmings, as well as food content in
crabs. It is also illustrated how intensity and propagation of
light-induced oxidation in dairy products and cod caviar can be done by
use of autofluorescence spectroscopy. Different applications need
different multivariate calibration and sampling approaches, and in this
talk this will be emphasised.
I kvantitativ tolkning av empiriske data kan uventede fenomener (interferenser) forårsake feilaktige resultater og tolkninger. Men for multirespons-data kan ukjente interferenser bli oppdaget, beskrevet og korrigert for ved hjelp av multivariat analyse av residualtabeller. Imidlertid er det en merkverdig begrensning i dette, som jeg har kalt paradokset om ” Den Informative Konverse ” (The Informative Converse) . Det har følgende konsekvens: Ufullstendig bakgrunnskunnskap kan låse vår forståelse og vår umiddelbare tolking av data. Men ved multivariat residualanalyse kan man få informasjon om de uventede interferensene, av en type som er ”konvers” i forhold til bakgrunnskunnskapen. Det kan således vises matematisk at det vi vet mest om på forhånd, kan vi lære minst om:
1) Bruker vi rekkevis bakgrunnskunnskap til analyse av en kjemisk datatabell, og denne bakgrunnskunnskapen er utilstrekkelig, vil vi KUN få riktig kolonnevis informasjon om de uventede interferensene. Anta at vi for eksempel bruker vanlig variansanalyse eller multirespons regresjon for et sett målevariabler Y (n x q), for eksempel absorbans ved q spektrale bølgelengder i n objekter. Anta videre at vi gjør dette med bruk av ufullstendig kunnskap: Vi har bare et subsett av p kjente forklaringsvariabler X (n x p) i n objekter (for eksempel design, eller kjente konsistuent-konsentrasjoner), mens andre, viktige men uventede variasjonsfenomener utelatt. Vi bruker den vanlige linerærmodellen Y=XB+F for å estimere effektene i B(p x q) og deres tilsynelatende ”signifikans”. Da vil uventede og følgelig ikke-modellerte effekter i Y kunne gi feilaktige effekt-estimater av B i variansanalysen eller regresjonen (”aliasfeil”). Dette fenomenet kan ha negative konsekvenser for tolkning av variansanalyser og regresjonsanalyser i systemer med uventede, systematiske feil. Ved etterfølgende multivariatanalyse av tabellen av de umodellerte residualene F kan vi imidlertid få ut riktig informasjon om noen (men ikke alle) aspekter av de uventede interferenseffektene: Vi kan nemlig få riktig estimat av interferensenes q-dimensjonale profiler (for eksempel deres spektra), men IKKE deres konsentrasjoner i de n objektene. Denne residualanalysen kan gi ny innsikt om uventede men viktige fenomener i ens systemer. Multivariat kalibrering (Martens & Næs 1989)[1] korrigerer for slike uventede interferens-effekter automatisk.
2) Bruker vi i stedet kolonnevis bakgrunnskunnskap til analyse av en kjemisk datatabell, og denne bakgrunnskunnskapen er utilstrekkelig, vil vi KUN få riktig rekkevis informasjon om de uventede interferensene. Anta for eksempel at et sett av n blandinger, hver karakterisert ved q målevariabler i Y (n x q), blir modellert additivt som kjemiske blandinger – d.v.s. som lineærkombinasjoner av p kjente konstituenters q-dimensjonale profiler S(p x q) (for eksempel analytenes spektra). Hvis den lineære blandingsmodellen Y=CS’ + F blir brukt til å projisere blandingsdata Y på analytspektra S for å estimere analytkonsentrasjonene C(n x p), vil uventede og følgelig ikke-modellerte fenomener i Y kunne ødelegge disse konsentrasjonsestimatene C, især hvis spektrene til disse ukjente fenomener ligner på de kjente spektrene til analytene, S. Dette kan ha negative konsekvenser for alle de teknikkene i for eksempel moderne fluorokrom-basert genomisk analyse, der man kun måler det antall bølgelengder lys som tilsvarer det antall fluorokromer man vet at man har tilsatt (typisk 2, hvis man bruker cy-3 og cy-5). Eventuelle uventede og ukjente interferenseffekter pga fluorescerence forurensninger osv vil da kunne gi store tolkningsfeil. Fra tabellen av umodellerte residualer F kan man imidlertid få ut noe (men ikke all) informasjon om de uventede interferenseffektene – nemlig kun konsentrasjonene deres i de n prøvene – men IKKE profilene deres q-dimensjonale spektra. Hadde man for eksempel målt fluorescens ved flere bølgelengder, kunne man ha oppdaget fordelingen av slike evt. uventede interferensfenomener. Dermed kunne man ha oppdaget hvor forurensningen kom fra, og således forbedret analyseteknikken ved neste korsvei, for eksempel ved endre laboratorierutinene. Hadde man hastverk, kunne man alternativt ganske enkelt brukt multivariat kalibrering til automatisk å estimere og kompensere for slike uidentifiserte interferenser.
Disse to “vinduene inn i det ukjente”,
beskrevet i
Martens (2011)[2], vil bli forklart matematisk. Så vil de bli
illustrert for absorbansspektra fra pulverblandinger, for å
demonstrere problemer med ufullstendig deduktiv kausalitetstolkning,
samt muligheten av etterfølgende induktiv oppdagelse og
kvantitativ beskrivelse av uventede fenomener. Paradokset vil
også bli diskutert epistemologisk, for de har muligens noe
med
hvordan forståelse og kultur utvikler seg generelt.
Referanser:
BigDFT is an ab initio pseudopotential DFT code based on Daubechies wavelets. They are an orthonormal compact support multiresolution basis, and form one of the few examples of systematic real space basis sets. For these reasons they provide good efficiency in expanding localized information. An ab initio code based on wavelets is thus optimal to calculate electronic properties of big inhomogeneous systems such as found in nanoscience or biology and for different boundary conditions (isolated systems, surface,...). At present, a stable and robust version of such code is ready, distributed with GNU-GPL license and integrated in ABINIT v. 5.5.x. The systematicity of the basis set together with the massive optimization of the operations performed make this code to be very fast and precise, with excellent efficiency on parallel and hybrid computers. The properties of the basis function are also well-suited for developing an O(N) code, which is one of the perspectives. During this presentation we will present the main features of the code, some applications using the capabilities of BigDFT and the present developments.
Catalysts are important in order to obtain environmentally friendly, less energy-consuming and cleaner chemical processes, and the majority of biological and chemical processes are catalyzed. It is thus not surprising that catalysis represents an important application area for molecular-level computational studies. Traditionally the most important role of such computational investigations has been to provide insight into the mechanisms with which existing catalysts mediate chemical processes. In recent years, however, the examples in which theory and molecular-level computational studies are used directly in prediction and development of new catalysts have become more frequent, and some examples of such theory-assisted design will be presented. The design methods thus covered will range from quantitative structure-activity relationships (QSAR) based on quantum chemically obtained molecular descriptors to a recently developed de novo evolutionary algorithm for automated building and optimization of homogeneous catalysts.
Electrically insulating materials have the role of isolating a
conductor either from another conductor or from ground. An insulating
material therefore continuously experiences an applied voltage, however
electrical breakdown properties are better discussed in terms of the
electric field in the insulating material. It is the electric field
strength at which the insulating properties of the material break down,
termed the dielectric
strength, that is the property of interest.
From a molecular perspective, several kinds of processes may occur at
high electric fields as for example ionization of molecules, creation
of anions by attachment of electrons, and molecular dissociation into
fragments. The initiation of breakdown requires a
“free” electron which is accelerated by the
electric field. If it has obtained sufficient kinetic energy when it
collides with a molecule, it may ionize the molecule and create another
“free” electron. Eventually, a plasma of electrons
and atomic and molecular fragments, termed a a streamer, is propagated
through the material driven by the electric field.
In an initial study [1], we therefore studied the electric-field
dependence on the ionization potential (IP) for typical molecules used
for liquid insulation either as base liquids or additives. The
calculation of the field-dependent IP is not trivial since the energy
reaches minus infinity at infinite separation between the cation and
the electron. An approximate method, based on studying the potential
surface of the interaction between a cation and a negative point charge
in an electric field, was developed to address this problem.
A main result is that the IP has a strong field dependence, and for the
molecules included in our study much stronger than the field dependence
on the excitation energies. Consequently, at a given field, well within
the region of an insulating liquid, a two-state model is obtained with
the electronic ground state and the ionized state. These insights have
given rise to several new ideas of which molecular processes that are
important and how a material for electrical insulation should be
designed.
The simplest approach to calculate total energies from
diagrammatic techniques is the random phase approximation (RPA) first
suggested and applied by Nozieres and Pines. With the tremendous
improvements in computer performance and using efficient
implementations, we are now able to apply the RPA to fairly large
systems. In this talk, I present a survey of our recent results,
covering lattice constants, bulk moduli, and atomisation energies of
prototypical solids[1-2]. The results are generally significantly
improved over conventional density functional theory calculations.
Specifically, we demonstrate that predicted equilibrium volumes of
alkali and alkali earth metals, transition metals and noble metals are
within 1-2 percent of experiment.
Setting out from this observation we apply the method to more
challenging problems, such as surface energies, adsorption energies of
small molecules on surfaces [3], and van der Waals bonded systems
(graphene) [4]. The results suggest that the RPA outperforms in many
respects available density functionals, accounting equally well for van
der Waals bonding, ionic, covalent, and metallic bonding. However, a
slight tendency towards underbinding is observed, making accurate
predictions (chemical accuracy) not yet possible. Possible solutions to
this problem, such as second order perturpation theory (MP2) and
coupled cluster methods, are briefly discussed.
References
A key concept in chemistry is the chemical bond and the
concept of sharing of electron pairs. Many debates in the chemical
literature are related to the understanding of the chemical bond and
how atoms are held together through electron pair sharing. In this
talk, I will present a new way of visualizing the chemical bond and the
sharing of electron pairs through the visualization of magnetically
induced currents. These currents allow us to visualize the flow of
electrons between bonded atoms in molecules, and also the repulsion or
lack of bonding between atoms.
I will present a series of calculations of magnetically induced
currents in different bonding situations (single bond, double bond,
triple bonds, hydrogen bonds, van der Waals interactions) as a function
of bond length and in particular focus on the change in the
magnetically-induced currents as we gradually break the chemical bond.
I will show that these magnetically-induced current may help us
understand the chemical bond in unconventional bonding
situations.
Formaldehyde [1] and glycoaldehyde [2] have both been observed
in interstellar environments, and a tentative detection of the next
higher carbohydrate, glyceraldehyde, has been reported. In the formose
reaction, which has been known since 1861,[3] formaldehyde oligomerizes
under basic conditions in water solution to give a mixture of
carbohydrates, formally:
AbstractLiving systems in nature are mostly constructed from carbon based polymeric structures and their composites with inorganic molecules. Such as; soft tissue is mostly formed from collagen, and hard tissue has collagen-hydroxyapatite composites. In labs, carbon based polymers can be prepared in different chemical and physical forms with various thermal and mechanical properties depending on the desire, and therefore, they are the preferred materials to be used in medicine.
In medical applications, artificial polymers are used in the production of catheters, heart valves, veins, contact lenses, etc; biodegradable and biocompatible natural originated polymers are the preferred ones used for the preparation of scaffolds as well as micro and nano capsules or hydrogels for the production of drug carriers. It is known that whenever an implant is replaced in the body, the first reactions take place on the surface. In some cases, although the synthesized material has the desired properties, the chemistry or physical form of the surface may not be compatible with the biological media. Therefore, some modifications of the medical devices and scaffolds are needed to enhance the biocompatibility of the system.
The materials that are used for tissue engineering purposes
can be
modified chemical or physical processes, or by radiation. Glow charge
plasma in the presence of an active or inert gas or by further
processes by binding other molecules covalently to the surface after
plasma activation, is one commonly applied technique. Surface of the
materials can be activated by glow discharge and the surface
hydrophilicity and surface free energy (SFE) can be changed either in
hydrophilic or hydrophobic direction. It was shown that, PMMA and PLLA
surfaces modified with oxygen plasma had the highest cell attachment
when the SFE values were about 60 mJ/m2. 1-3 It was also shown that
combination of antithrombogenic molecules like heparin to the surfaces
after plasma activation increased their antithrombogenic property.4
Addition of nanoparticles either on or in of the fibrous matrices and
loading them with growth factors increased their bioactivity in the
formation of new tissues in bone tissue regeneration.5-7 Similarly
addition of micro-nano sized hydroxyapatite (HAp) particles made the
surfaces more bioactive and osteoconductive, and enhanced the
attachment of osteoblast cells to the surface, while addition of
antibacterial molecules or systems containing antibacterial property
prevented the attachment of bacteria.8
In this presentation some examples related to the preparations of
polymeric composites and some nano-micro modifications as given below,
will be discussed.
References
Recent interest in stimuli-sensitive materials has promoted numerous efforts in preparing “intelligent” hydrogels [1,2]. Hydrogels are viscoelastic materials constituted of three dimensional and continuous networks formed by polymers and water molecules entrapped within the networks. The “smart” hydrogels have various potential applications in biomedical materials field, especially in controlled drug delivery systems [3]. There are many kinds of stimuli-responsive hydrogels that can response to the external alterations in environmental conditions such as temperature, pH, and photo. Polysaccharides and other biopolymers, with or without co-solutes, are frequently utilized as responsive polymers, and we have focused on these systems.
Stimuli-sensitive nano- or microgels are polymeric particles that consist of cross-linked three-dimensional networks. They shrink or swell significantly by expelling or absorbing large amounts of water in response to external changes in temperature, pH, or magnetic fields. The chemical composition of the nano-gel determines the stimulus that can trigger the volume-phase transition. The dramatic response and stimuli-specific behavior makes these materials extremely valuable for numerous applications, including drug delivery [4]. We will illustrate this swelling-deswelling effect by some examples.
Amphiphilic block copolymers having both hydrophilic and hydrophobic segments are known to form micellar structures in aqueous media due to their amphiphilic character [5].Highly hydrated outer shells of polymeric micelles can inhibit intermicellar aggregation of hydrophobic inner cores. Consequently, polymeric micelles maintain their satisfactory aqueous stability irrespective of the high content of hydrophobic drug bound within the micelle inner core.
Nanocomposite materials consisting of colloidal metal (e.g., gold) nanoparticles covered by a layer of a synthetic polymer have attracted attention as targeting “bullets” in drug delivery applications. Some special features of these nanoparticles will be shown.
Dendrimers are highly branched, globular macromolecules with many
arms emanating from a central core [6]. The stepwise synthesis of
dendrimers affords molecules with a highly regular branching pattern, a
unique molecular weight or a low polydispersity index, and a
well-defined number of peripheral groups. Dendrimers have potential as
macromolecular vectors in novel drug delivery and biomedical
applications. A few simple examples will be shown.
References
Otto Wichterle pioneered the use of hydrogels for
manufacturing soft
contact lenses with his groundbreaking invention in 1960 of using
2-hydroxyethyl-methacrylate (HEMA) as monomer to generate polyHEMA by
radical polymerization. Before that, contact lenses were manufactured
from rather stiff materials like glass or PMMA. Besides good optical
performance, the polyHEMA lenses provided vastly more comfort to the
wearer. After that most of contact lens R&D work was invested
in
modifying the crosslinked hydrogels in direction of improved
materials’ properties providing better comfort and
biocompatibility as well as better economics and higher optical
quality, which finally allowed the introduction of daily disposable
contact lenses, eliminating the inconvenience of cleaning and storing
the lenses, and also avoiding any problems with deposits from tear
components on or in the lens.
A major unmet need of conventional hydrogels was the limited oxygen
permeability, not being able to always provide the necessary oxygen to
the cornea, specifically with closed eyes. Continuous overnight wear of
contact lenses would provide another dimension of convenience and
health, avoiding the daily handling and cleaning of lenses for a week,
a month or so, and allowing the wearer to wake up in the middle of the
night or in the morning with immediate corrected vision. But
insufficient oxygen supply to the cornea, or hypoxia, provokes a
moderate swelling of the cornea, which is usually totally reversible
after allowing sufficient oxygen to reach the cornea. However, longer
term hypoxia leads to red eyes because the hypoxic cornea signals this
condition, which triggers blood vessels to grow from the sclera to the
cornea.
The oxygen permeability of a conventional hydrogel is controlled
essentially by its water content. But lenses made from materials even
with high water content do not supply sufficient oxygen for healthy
overnight wear. Although providing sufficient oxygen to the cornea,
contact lenses from silicone-containing acrylates or even pure
silicones suffer from distinct disadvantages with respect to
wettability, discomfort, and the tendency to stick onto the cornea
– disadvantages mainly due to the lack of water in the
polymeric
material, not allowing any ion transport. Only a suitable combination
of a hydrogel and a silicone type material fulfills both requirements
for water/ion transport and high gas (oxygen) permeability. This was
achieved with a bi-phasic network with distinctly different hydrogel
and silicone phases, whereby a co-continuous morphology is necessary
for good clinical performance. Thus, both phases in the lens material
provide each a continuous transport pathway from the front to the back
side of the contact lens. This was realized by the correct choice of
the polymer structure and block lengths of the constituents. Optical
clarity of such bi-phasic materials can be maintained by keeping the
domain size sufficiently below the wavelength of visible light.
During the past decade, conventional surface plasmon resonance (SPR) has started to play a central role in the drug screening process. Recently, the through-put capacity of these systems has increased by integrating array-based SPR systems with imaging readout and advanced microfluidic liquid handling. However, what still remains a holy grail in this line of research is the limited success of SPR to probe the interaction of drug candidates with lipid-membrane-residing proteins. While water-soluble proteins can be immobilized at sufficiently high densities to probe the binding even of low-molecular-weight drug candidates, the fact that lipids must surround membrane proteins puts sever constrains on the surface densities, and thus signal to noise ratios, that can be reached while still keeping the proteins in a native state. With 30% of all membrane proteins acting as molecular channels, and up to 70% of all current drugs being targeted towards membrane proteins, there is a need for new solutions to this challenge. Emerging innovations are expected to require both new interfacial lipid-based self assembly approaches and surface-based transducer concepts with improved sensitivity. In this presentation, I will discuss our recent attempts to use evanescent wave sensing to probe membrane-protein controlled molecular transport,[1] as well as our recent efforts to merge small-scale LSPR sensing[2] with liquid handling providing millisecond solution-exchange rates over micron sized areas as well as means to control the motion and position of supported lipid bilayers using hydrodynamic flows.[3] I will also present how LSPR-active nanoholes can be utilized as substrate-spanning pores, thus enabling flow-through sensing offering significantly improved rates of binding as well as capture efficiencies,[4] especially if combined with material-specific-surface chemistries that direct the molecular recognition reactions to the most sensitive regions of the LSPR sensors.[5] We have also explored total internal reflection fluorescence microscopy (TIRFM) to probe the kinetics of single biomolecular binding events[6] which combined with capacity of time-of-flight (TOF) secondary ion mass spectrometry (SIMS) to identify lipid vesicles with different lipid composition has also open up a new means towards multiplexed sensing on the level of single biorecognition reactions[7].
References
Transplantation of living cells is a potential strategy for treatment of various diseases caused by the body’s inability to produce required amounts of an essential molecule such as a hormone or an enzyme. Such cell therapy treatment involves the replacement or repair of damaged tissue or cells by means of transplantation of human or animal cells. As for whole organ transplants, patients undergoing cell therapy treatments run the risk of rejection, in which the body recognizes the cells as a foreign substance and seeks to destroy the transplant.
Biopolymer microcapsules of alginate can be used as immune
barriers
for cell transplantation where the alginate gel protects the transplant
from the host immune system. The general concept of immunoisolation is
to prevent rejection by separating the transplanted cells from the
hostile immunological environment in the host by a semi-permeable
artificial membrane.
The far most studied cell therapy system is the encapsulation of insulin producing cells (islets of Langerhans) into calcium-alginate microcapsules for transplantation into diabetic patients as an approach to treat Type 1 diabetes. However, there are still some issues that remain to be solved before microencapsulated islets can be transferred to the clinic.
Despite numerous important methodological advancements in all areas of chemistry, still most organic synthesis as well as the industrial production of chemicals can be improved. Currently, more than 80% of all products of the chemical industry are made via catalysis. In this regard, the development of new and more efficient catalysts constitutes a key factor for achieving a sustainable production of all kinds of chemicals today and in the future. In the talk it will be shown that recently developed molecular-defined as well as nano-structured iron catalysts enable academic chemists to perform their organic syntheses with high yields and selectivities. In the future, also for industrial processes improved economics might be expected. Examples which demonstrate the superiority of catalytic processes with bio-relevant metal complexes compared to more traditional catalytic reactions will include hydrogenations and dehydrogenations, and other redox reactions. In addition, the need for drastically improved catalysts for challenging “dream reactions” in the area of environmentally benign energy technologies will be highlighted.
Our research interests include the development of controlled polymer architectures,1 hybrid nanoparticle assemblies,2 and the integration of dynamic supramolecular systems onto surfaces.3 Using cucurbit[n]urils (CB[n]s) we adopt a simple bottom-up approach to achieve sophisticated designs which are directed at the preparation of novel photonic devices, high-density patterned media, and chemical and biological sensors.4 Our CB[n] based host-guest systems exhibit dynamic self assembly and are capable of responding to stimuli (photochemical, chemical, and thermal) which allow for external control and function to be built into the materials. Modification of solution viscosity using multivalent polymers5 and imidazolium based ionic liquids6 have been accomplished through dynamic crosslinking in water using CB[n]s to produce colorful hydrogels and hierarchical architectures. Furthermore, polymer-inorganic composite materials can be readily prepared based on the CB[8] coupling of multivalent gold nanoparticles to copolymers.7 When these systems are attached onto gold surfaces intricate control is achieved over the site-selective immobilization of colloids3 and peptides. This has great scope for the development of optical materials, self-assembled hybrid systems for catalysis, chemical sensors and biological separations.
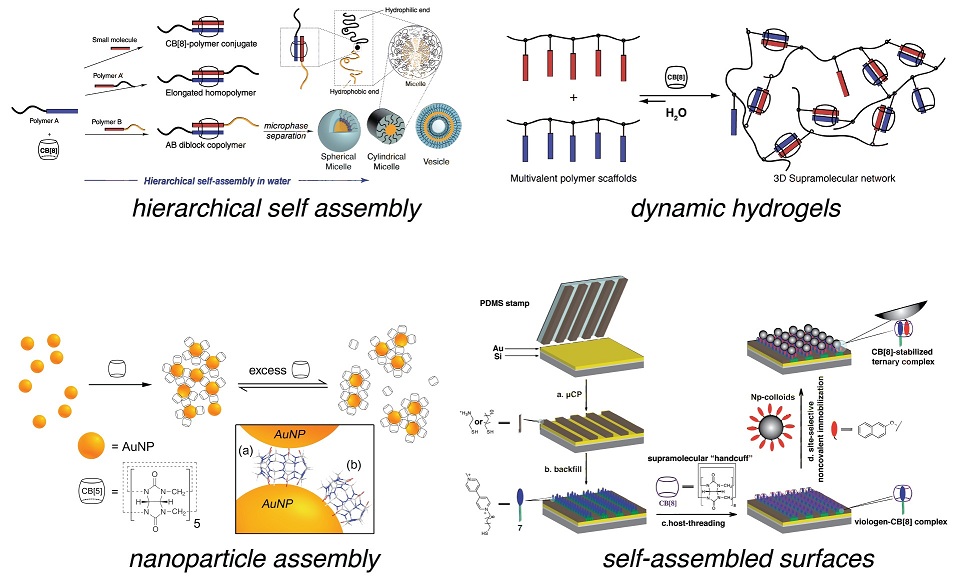
A flurry of recent activity has established that mixed-metal formulations which combine zinc with an alkali-metal are versatile organometallic reagents in synthesis, offering better chemo- and regio-selectivities than conventional monometallic reagents. Despite a large number of synthetic organic studies having utilising these reagents, the key factors governing their reactions still remain to be elucidated and fully understood.
We have approached this intriguing area from more of a metal-focussed perspective. This talk will present results from our work that provide new insights into the true identities of the organometallic intermediates involved in these reactions prior to any electrophilic interception step focussing on deprotonation,1 metal-halogen exchange2 and nucleophilic addition3 processes. By using a combination of X-ray crystallography, NMR spectroscopy and DFT calculations, we will discuss how the structures of, and the bonding within these isolated metallo intermediates, can help to rationalise the unique region and chemo-selectivities observed in these fundamental organic transformations.
References
Gold(I) complexes are the catalysts of choice for the selective
activation of alkynes under mild conditions, which has been applied for
the construction of complex cyclic systems.1 Thus, we have used the
gold(I)-catalyzed cyclization of 1,6-enynes with carbonyl groups at the
alkenyl chain for the synthesis of the sesquiterpenes orientalol F and
englerin A.2,3 Recently we have found that the intermolecular reaction
of alkynes with alkenes leads to cyclobutenes in the presence of
cationic gold(I) catalysts bearing bulky phosphine ligands.4 Extension
of these results to the development of new gold(I)-catalyzed
cyclization cascades will be presented.
References